Anna Zhukova, Frédéric Hecht, Yvon Maday, Olivier Gascuel
{"title":"从系统发生树快速、准确地估计多类型出生-死亡流行病学模型的最大似然法","authors":"Anna Zhukova, Frédéric Hecht, Yvon Maday, Olivier Gascuel","doi":"10.1093/sysbio/syad059","DOIUrl":null,"url":null,"abstract":"<p><p>Multi-type birth-death (MTBD) models are phylodynamic analogies of compartmental models in classical epidemiology. They serve to infer such epidemiological parameters as the average number of secondary infections Re and the infectious time from a phylogenetic tree (a genealogy of pathogen sequences). The representatives of this model family focus on various aspects of pathogen epidemics. For instance, the birth-death exposed-infectious (BDEI) model describes the transmission of pathogens featuring an incubation period (when there is a delay between the moment of infection and becoming infectious, as for Ebola and SARS-CoV-2), and permits its estimation along with other parameters. With constantly growing sequencing data, MTBD models should be extremely useful for unravelling information on pathogen epidemics. However, existing implementations of these models in a phylodynamic framework have not yet caught up with the sequencing speed. Computing time and numerical instability issues limit their applicability to medium data sets (≤ 500 samples), while the accuracy of estimations should increase with more data. We propose a new highly parallelizable formulation of ordinary differential equations for MTBD models. We also extend them to forests to represent situations when a (sub-)epidemic started from several cases (e.g., multiple introductions to a country). We implemented it for the BDEI model in a maximum likelihood framework using a combination of numerical analysis methods for efficient equation resolution. Our implementation estimates epidemiological parameter values and their confidence intervals in two minutes on a phylogenetic tree of 10,000 samples. Comparison to the existing implementations on simulated data shows that it is not only much faster but also more accurate. An application of our tool to the 2014 Ebola epidemic in Sierra-Leone is also convincing, with very fast calculation and precise estimates. As MTBD models are closely related to Cladogenetic State Speciation and Extinction (ClaSSE)-like models, our findings could also be easily transferred to the macroevolution domain.</p>","PeriodicalId":22120,"journal":{"name":"Systematic Biology","volume":" ","pages":"1387-1402"},"PeriodicalIF":5.7000,"publicationDate":"2023-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10924745/pdf/","citationCount":"0","resultStr":"{\"title\":\"Fast and Accurate Maximum-Likelihood Estimation of Multi-Type Birth-Death Epidemiological Models from Phylogenetic Trees.\",\"authors\":\"Anna Zhukova, Frédéric Hecht, Yvon Maday, Olivier Gascuel\",\"doi\":\"10.1093/sysbio/syad059\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Multi-type birth-death (MTBD) models are phylodynamic analogies of compartmental models in classical epidemiology. They serve to infer such epidemiological parameters as the average number of secondary infections Re and the infectious time from a phylogenetic tree (a genealogy of pathogen sequences). The representatives of this model family focus on various aspects of pathogen epidemics. For instance, the birth-death exposed-infectious (BDEI) model describes the transmission of pathogens featuring an incubation period (when there is a delay between the moment of infection and becoming infectious, as for Ebola and SARS-CoV-2), and permits its estimation along with other parameters. With constantly growing sequencing data, MTBD models should be extremely useful for unravelling information on pathogen epidemics. However, existing implementations of these models in a phylodynamic framework have not yet caught up with the sequencing speed. Computing time and numerical instability issues limit their applicability to medium data sets (≤ 500 samples), while the accuracy of estimations should increase with more data. We propose a new highly parallelizable formulation of ordinary differential equations for MTBD models. We also extend them to forests to represent situations when a (sub-)epidemic started from several cases (e.g., multiple introductions to a country). We implemented it for the BDEI model in a maximum likelihood framework using a combination of numerical analysis methods for efficient equation resolution. Our implementation estimates epidemiological parameter values and their confidence intervals in two minutes on a phylogenetic tree of 10,000 samples. Comparison to the existing implementations on simulated data shows that it is not only much faster but also more accurate. An application of our tool to the 2014 Ebola epidemic in Sierra-Leone is also convincing, with very fast calculation and precise estimates. As MTBD models are closely related to Cladogenetic State Speciation and Extinction (ClaSSE)-like models, our findings could also be easily transferred to the macroevolution domain.</p>\",\"PeriodicalId\":22120,\"journal\":{\"name\":\"Systematic Biology\",\"volume\":\" \",\"pages\":\"1387-1402\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2023-12-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10924745/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Systematic Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/sysbio/syad059\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"EVOLUTIONARY BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Systematic Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/sysbio/syad059","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
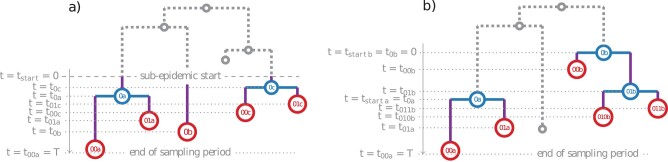
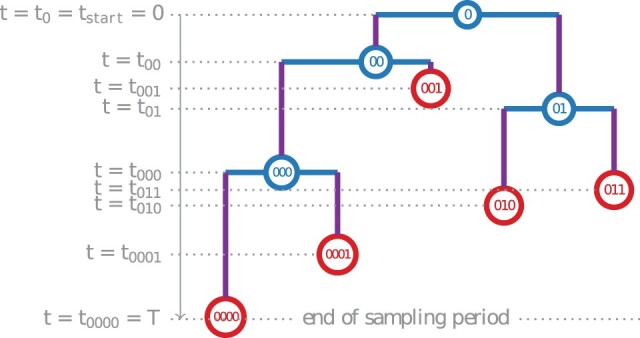
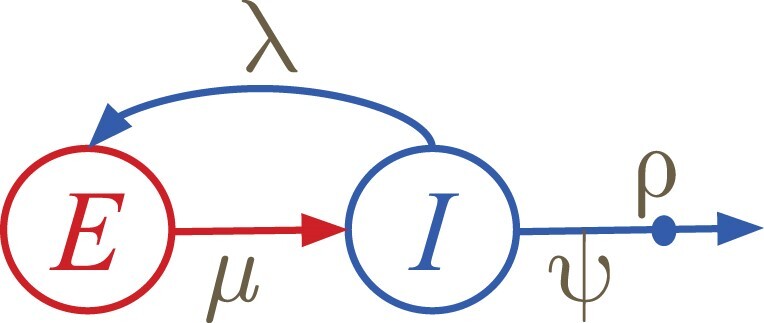
多类型出生-死亡(MTBD)模型是经典流行病学中分区模型的系统动力学类似物。它们可以从系统发育树(病原体序列的系谱)中推断出二次感染的平均数量和感染时间等流行病学参数。该模型系列的代表侧重于病原体流行的各个方面。例如,"出生-死亡-暴露-感染"(BDEI)模型描述了具有潜伏期的病原体传播(从感染到具有传染性之间存在延迟,如埃博拉病毒和 SARS-CoV-2),并允许与其他参数一起进行估算。随着测序数据的不断增加,MTBD 模型对于揭示病原体流行信息应该非常有用。然而,这些模型在系统动力学框架下的现有实现还跟不上测序速度。计算时间和数值不稳定性问题限制了它们对中等数据集(≤ 500 个样本)的适用性,而估计的准确性应随着数据量的增加而提高。我们为 MTBD 模型提出了一种新的高度可并行化的常微分方程公式。我们还将其扩展到森林,以表示一种(亚)流行病从多个病例开始的情况(如一个国家的多次引入)。我们在最大似然法框架内对 BDEI 模型实施了这一方法,并结合使用了数值分析方法来有效解决方程问题。我们的实施方案能在两分钟内估算出包含 10,000 个样本的系统发生树上的流行病学参数值及其置信区间。与现有的模拟数据实施方案相比,我们的实施方案不仅速度更快,而且更加准确。我们的工具在 2014 年塞拉利昂埃博拉疫情中的应用也令人信服,计算速度非常快,估计值也很精确。由于 MTBD 模型与类似于 Cladogenetic State Speciation and Extinction(ClaSSE)的模型密切相关,我们的发现也可以很容易地转移到宏观进化领域。
Fast and Accurate Maximum-Likelihood Estimation of Multi-Type Birth-Death Epidemiological Models from Phylogenetic Trees.
Multi-type birth-death (MTBD) models are phylodynamic analogies of compartmental models in classical epidemiology. They serve to infer such epidemiological parameters as the average number of secondary infections Re and the infectious time from a phylogenetic tree (a genealogy of pathogen sequences). The representatives of this model family focus on various aspects of pathogen epidemics. For instance, the birth-death exposed-infectious (BDEI) model describes the transmission of pathogens featuring an incubation period (when there is a delay between the moment of infection and becoming infectious, as for Ebola and SARS-CoV-2), and permits its estimation along with other parameters. With constantly growing sequencing data, MTBD models should be extremely useful for unravelling information on pathogen epidemics. However, existing implementations of these models in a phylodynamic framework have not yet caught up with the sequencing speed. Computing time and numerical instability issues limit their applicability to medium data sets (≤ 500 samples), while the accuracy of estimations should increase with more data. We propose a new highly parallelizable formulation of ordinary differential equations for MTBD models. We also extend them to forests to represent situations when a (sub-)epidemic started from several cases (e.g., multiple introductions to a country). We implemented it for the BDEI model in a maximum likelihood framework using a combination of numerical analysis methods for efficient equation resolution. Our implementation estimates epidemiological parameter values and their confidence intervals in two minutes on a phylogenetic tree of 10,000 samples. Comparison to the existing implementations on simulated data shows that it is not only much faster but also more accurate. An application of our tool to the 2014 Ebola epidemic in Sierra-Leone is also convincing, with very fast calculation and precise estimates. As MTBD models are closely related to Cladogenetic State Speciation and Extinction (ClaSSE)-like models, our findings could also be easily transferred to the macroevolution domain.
期刊介绍:
Systematic Biology is the bimonthly journal of the Society of Systematic Biologists. Papers for the journal are original contributions to the theory, principles, and methods of systematics as well as phylogeny, evolution, morphology, biogeography, paleontology, genetics, and the classification of all living things. A Points of View section offers a forum for discussion, while book reviews and announcements of general interest are also featured.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


