Amy Moore, Mitchell J Machiela, Moara Machado, Sophia S Wang, Eleanor Kane, Susan L Slager, Weiyin Zhou, Mary Carrington, Qing Lan, Roger L Milne, Brenda M Birmann, Hans-Olov Adami, Demetrius Albanes, Alan A Arslan, Nikolaus Becker, Yolanda Benavente, Simonetta Bisanzi, Paolo Boffetta, Paige M Bracci, Paul Brennan, Angela R Brooks-Wilson, Federico Canzian, Neil Caporaso, Jacqueline Clavel, Pierluigi Cocco, Lucia Conde, David G Cox, Wendy Cozen, Karen Curtin, Immaculata De Vivo, Silvia de Sanjose, Lenka Foretova, Susan M Gapstur, Hervè Ghesquières, Graham G Giles, Martha Glenn, Bengt Glimelius, Chi Gao, Thomas M Habermann, Henrik Hjalgrim, Rebecca D Jackson, Mark Liebow, Brian K Link, Marc Maynadie, James McKay, Mads Melbye, Lucia Miligi, Thierry J Molina, Alain Monnereau, Alexandra Nieters, Kari E North, Kenneth Offit, Alpa V Patel, Sara Piro, Vignesh Ravichandran, Elio Riboli, Gilles Salles, Richard K Severson, Christine F Skibola, Karin E Smedby, Melissa C Southey, John J Spinelli, Anthony Staines, Carolyn Stewart, Lauren R Teras, Lesley F Tinker, Ruth C Travis, Claire M Vajdic, Roel C H Vermeulen, Joseph Vijai, Elisabete Weiderpass, Stephanie Weinstein, Nicole Wong Doo, Yawei Zhang, Tongzhang Zheng, Stephen J Chanock, Nathaniel Rothman, James R Cerhan, Michael Dean, Nicola J Camp, Meredith Yeager, Sonja I Berndt
{"title":"四种非霍奇金淋巴瘤亚型的全基因组纯合性和风险。","authors":"Amy Moore, Mitchell J Machiela, Moara Machado, Sophia S Wang, Eleanor Kane, Susan L Slager, Weiyin Zhou, Mary Carrington, Qing Lan, Roger L Milne, Brenda M Birmann, Hans-Olov Adami, Demetrius Albanes, Alan A Arslan, Nikolaus Becker, Yolanda Benavente, Simonetta Bisanzi, Paolo Boffetta, Paige M Bracci, Paul Brennan, Angela R Brooks-Wilson, Federico Canzian, Neil Caporaso, Jacqueline Clavel, Pierluigi Cocco, Lucia Conde, David G Cox, Wendy Cozen, Karen Curtin, Immaculata De Vivo, Silvia de Sanjose, Lenka Foretova, Susan M Gapstur, Hervè Ghesquières, Graham G Giles, Martha Glenn, Bengt Glimelius, Chi Gao, Thomas M Habermann, Henrik Hjalgrim, Rebecca D Jackson, Mark Liebow, Brian K Link, Marc Maynadie, James McKay, Mads Melbye, Lucia Miligi, Thierry J Molina, Alain Monnereau, Alexandra Nieters, Kari E North, Kenneth Offit, Alpa V Patel, Sara Piro, Vignesh Ravichandran, Elio Riboli, Gilles Salles, Richard K Severson, Christine F Skibola, Karin E Smedby, Melissa C Southey, John J Spinelli, Anthony Staines, Carolyn Stewart, Lauren R Teras, Lesley F Tinker, Ruth C Travis, Claire M Vajdic, Roel C H Vermeulen, Joseph Vijai, Elisabete Weiderpass, Stephanie Weinstein, Nicole Wong Doo, Yawei Zhang, Tongzhang Zheng, Stephen J Chanock, Nathaniel Rothman, James R Cerhan, Michael Dean, Nicola J Camp, Meredith Yeager, Sonja I Berndt","doi":"10.20517/jtgg.2021.08","DOIUrl":null,"url":null,"abstract":"<p><strong>Aim: </strong>Recessive genetic variation is thought to play a role in non-Hodgkin lymphoma (NHL) etiology. Runs of homozygosity (ROH), defined based on long, continuous segments of homozygous SNPs, can be used to estimate both measured and unmeasured recessive genetic variation. We sought to examine genome-wide homozygosity and NHL risk.</p><p><strong>Methods: </strong>We used data from eight genome-wide association studies of four common NHL subtypes: 3061 chronic lymphocytic leukemia (CLL), 3814 diffuse large B-cell lymphoma (DLBCL), 2784 follicular lymphoma (FL), and 808 marginal zone lymphoma (MZL) cases, as well as 9374 controls. We examined the effect of homozygous variation on risk by: (1) estimating the fraction of the autosome containing runs of homozygosity (FROH); (2) calculating an inbreeding coefficient derived from the correlation among uniting gametes (F3); and (3) examining specific autosomal regions containing ROH. For each, we calculated beta coefficients and standard errors using logistic regression and combined estimates across studies using random-effects meta-analysis.</p><p><strong>Results: </strong>We discovered positive associations between FROH and CLL (β = 21.1, SE = 4.41, <i>P</i> = 1.6 × 10<sup>-6</sup>) and FL (β = 11.4, SE = 5.82, <i>P</i> = 0.02) but not DLBCL (<i>P</i> = 1.0) or MZL (<i>P</i> = 0.91). For F3, we observed an association with CLL (β = 27.5, SE = 6.51, <i>P</i> = 2.4 × 10<sup>-5</sup>). We did not find evidence of associations with specific ROH, suggesting that the associations observed with FROH and F3 for CLL and FL risk were not driven by a single region of homozygosity.</p><p><strong>Conclusion: </strong>Our findings support the role of recessive genetic variation in the etiology of CLL and FL; additional research is needed to identify the specific loci associated with NHL risk.</p>","PeriodicalId":73999,"journal":{"name":"Journal of translational genetics and genomics","volume":"5 ","pages":"200-217"},"PeriodicalIF":1.0000,"publicationDate":"2021-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8494431/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genome-wide homozygosity and risk of four non-Hodgkin lymphoma subtypes.\",\"authors\":\"Amy Moore, Mitchell J Machiela, Moara Machado, Sophia S Wang, Eleanor Kane, Susan L Slager, Weiyin Zhou, Mary Carrington, Qing Lan, Roger L Milne, Brenda M Birmann, Hans-Olov Adami, Demetrius Albanes, Alan A Arslan, Nikolaus Becker, Yolanda Benavente, Simonetta Bisanzi, Paolo Boffetta, Paige M Bracci, Paul Brennan, Angela R Brooks-Wilson, Federico Canzian, Neil Caporaso, Jacqueline Clavel, Pierluigi Cocco, Lucia Conde, David G Cox, Wendy Cozen, Karen Curtin, Immaculata De Vivo, Silvia de Sanjose, Lenka Foretova, Susan M Gapstur, Hervè Ghesquières, Graham G Giles, Martha Glenn, Bengt Glimelius, Chi Gao, Thomas M Habermann, Henrik Hjalgrim, Rebecca D Jackson, Mark Liebow, Brian K Link, Marc Maynadie, James McKay, Mads Melbye, Lucia Miligi, Thierry J Molina, Alain Monnereau, Alexandra Nieters, Kari E North, Kenneth Offit, Alpa V Patel, Sara Piro, Vignesh Ravichandran, Elio Riboli, Gilles Salles, Richard K Severson, Christine F Skibola, Karin E Smedby, Melissa C Southey, John J Spinelli, Anthony Staines, Carolyn Stewart, Lauren R Teras, Lesley F Tinker, Ruth C Travis, Claire M Vajdic, Roel C H Vermeulen, Joseph Vijai, Elisabete Weiderpass, Stephanie Weinstein, Nicole Wong Doo, Yawei Zhang, Tongzhang Zheng, Stephen J Chanock, Nathaniel Rothman, James R Cerhan, Michael Dean, Nicola J Camp, Meredith Yeager, Sonja I Berndt\",\"doi\":\"10.20517/jtgg.2021.08\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Aim: </strong>Recessive genetic variation is thought to play a role in non-Hodgkin lymphoma (NHL) etiology. Runs of homozygosity (ROH), defined based on long, continuous segments of homozygous SNPs, can be used to estimate both measured and unmeasured recessive genetic variation. We sought to examine genome-wide homozygosity and NHL risk.</p><p><strong>Methods: </strong>We used data from eight genome-wide association studies of four common NHL subtypes: 3061 chronic lymphocytic leukemia (CLL), 3814 diffuse large B-cell lymphoma (DLBCL), 2784 follicular lymphoma (FL), and 808 marginal zone lymphoma (MZL) cases, as well as 9374 controls. We examined the effect of homozygous variation on risk by: (1) estimating the fraction of the autosome containing runs of homozygosity (FROH); (2) calculating an inbreeding coefficient derived from the correlation among uniting gametes (F3); and (3) examining specific autosomal regions containing ROH. For each, we calculated beta coefficients and standard errors using logistic regression and combined estimates across studies using random-effects meta-analysis.</p><p><strong>Results: </strong>We discovered positive associations between FROH and CLL (β = 21.1, SE = 4.41, <i>P</i> = 1.6 × 10<sup>-6</sup>) and FL (β = 11.4, SE = 5.82, <i>P</i> = 0.02) but not DLBCL (<i>P</i> = 1.0) or MZL (<i>P</i> = 0.91). For F3, we observed an association with CLL (β = 27.5, SE = 6.51, <i>P</i> = 2.4 × 10<sup>-5</sup>). We did not find evidence of associations with specific ROH, suggesting that the associations observed with FROH and F3 for CLL and FL risk were not driven by a single region of homozygosity.</p><p><strong>Conclusion: </strong>Our findings support the role of recessive genetic variation in the etiology of CLL and FL; additional research is needed to identify the specific loci associated with NHL risk.</p>\",\"PeriodicalId\":73999,\"journal\":{\"name\":\"Journal of translational genetics and genomics\",\"volume\":\"5 \",\"pages\":\"200-217\"},\"PeriodicalIF\":1.0000,\"publicationDate\":\"2021-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8494431/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of translational genetics and genomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.20517/jtgg.2021.08\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/6/17 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of translational genetics and genomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.20517/jtgg.2021.08","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/6/17 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Genome-wide homozygosity and risk of four non-Hodgkin lymphoma subtypes.
Aim: Recessive genetic variation is thought to play a role in non-Hodgkin lymphoma (NHL) etiology. Runs of homozygosity (ROH), defined based on long, continuous segments of homozygous SNPs, can be used to estimate both measured and unmeasured recessive genetic variation. We sought to examine genome-wide homozygosity and NHL risk.
Methods: We used data from eight genome-wide association studies of four common NHL subtypes: 3061 chronic lymphocytic leukemia (CLL), 3814 diffuse large B-cell lymphoma (DLBCL), 2784 follicular lymphoma (FL), and 808 marginal zone lymphoma (MZL) cases, as well as 9374 controls. We examined the effect of homozygous variation on risk by: (1) estimating the fraction of the autosome containing runs of homozygosity (FROH); (2) calculating an inbreeding coefficient derived from the correlation among uniting gametes (F3); and (3) examining specific autosomal regions containing ROH. For each, we calculated beta coefficients and standard errors using logistic regression and combined estimates across studies using random-effects meta-analysis.
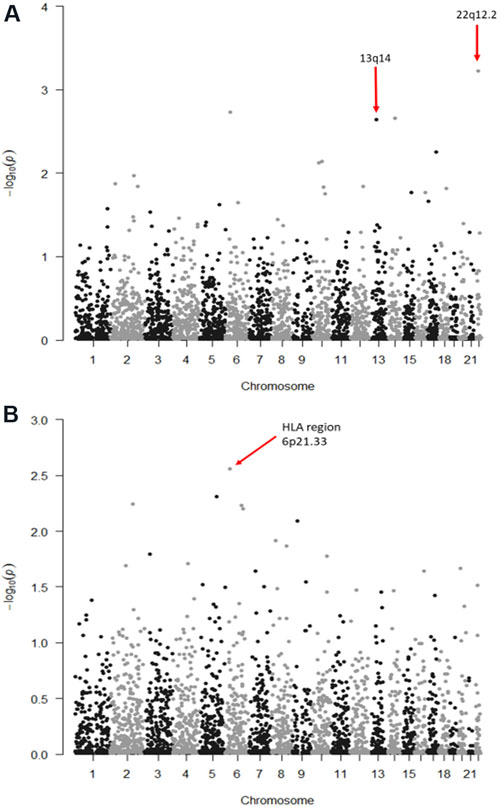
Results: We discovered positive associations between FROH and CLL (β = 21.1, SE = 4.41, P = 1.6 × 10-6) and FL (β = 11.4, SE = 5.82, P = 0.02) but not DLBCL (P = 1.0) or MZL (P = 0.91). For F3, we observed an association with CLL (β = 27.5, SE = 6.51, P = 2.4 × 10-5). We did not find evidence of associations with specific ROH, suggesting that the associations observed with FROH and F3 for CLL and FL risk were not driven by a single region of homozygosity.
Conclusion: Our findings support the role of recessive genetic variation in the etiology of CLL and FL; additional research is needed to identify the specific loci associated with NHL risk.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


