James Owen Delaluna, Heekyoung Kang, Yuan Yi Chang, MinJi Kim, Min-Ho Choi, Jun Kim, Hyun Beom Song
{"title":"使用基于纳米孔的长读测序技术对韩国个体的鞭虫进行从头组装的有丝分裂基因组分析。","authors":"James Owen Delaluna, Heekyoung Kang, Yuan Yi Chang, MinJi Kim, Min-Ho Choi, Jun Kim, Hyun Beom Song","doi":"10.1371/journal.pntd.0011586","DOIUrl":null,"url":null,"abstract":"<p><p>Knowledge about mitogenomes has been proven to be essential in human parasite diagnostics and understanding of their diversity. However, the lack of substantial data for comparative analysis is still a challenge in Trichuris trichiura research. To provide high quality mitogenomes, we utilized long-read sequencing technology of Oxford Nanopore Technologies (ONT) to better resolve repetitive regions and to construct de novo mitogenome assembly minimizing reference biases. In this study, we got three de novo assembled mitogenomes of T. trichiura isolated from Korean individuals. These circular complete mitogenomes of T. trichiura are 14,508 bp, 14,441 bp, and 14,440 bp in length. A total of 37 predicted genes were identified consisting of 13 protein-coding genes (PCGs), 22 transfer RNA (tRNAs) genes, two ribosomal RNA (rRNA) genes (rrnS and rrnL), and two non-coding regions. Interestingly, the assembled mitogenome has up to six times longer AT-rich regions than previous reference sequences, thus proving the advantage of long-read sequencing in resolving unreported non-coding regions. Furthermore, variant detection and phylogenetic analysis using concatenated protein coding genes, cox1, rrnL, and nd1 genes confirmed the distinct molecular identity of this newly assembled mitogenome while at the same time showing high genetic relationship with sequences from China or Tanzania. Our study provided a new set of reference mitogenome with better contiguity and resolved repetitive regions that could be used for meaningful phylogenetic analysis to further understand disease transmission and parasite biology.</p>","PeriodicalId":20260,"journal":{"name":"PLoS Neglected Tropical Diseases","volume":"17 8","pages":"e0011586"},"PeriodicalIF":3.4000,"publicationDate":"2023-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491297/pdf/","citationCount":"0","resultStr":"{\"title\":\"De novo assembled mitogenome analysis of Trichuris trichiura from Korean individuals using nanopore-based long-read sequencing technology.\",\"authors\":\"James Owen Delaluna, Heekyoung Kang, Yuan Yi Chang, MinJi Kim, Min-Ho Choi, Jun Kim, Hyun Beom Song\",\"doi\":\"10.1371/journal.pntd.0011586\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Knowledge about mitogenomes has been proven to be essential in human parasite diagnostics and understanding of their diversity. However, the lack of substantial data for comparative analysis is still a challenge in Trichuris trichiura research. To provide high quality mitogenomes, we utilized long-read sequencing technology of Oxford Nanopore Technologies (ONT) to better resolve repetitive regions and to construct de novo mitogenome assembly minimizing reference biases. In this study, we got three de novo assembled mitogenomes of T. trichiura isolated from Korean individuals. These circular complete mitogenomes of T. trichiura are 14,508 bp, 14,441 bp, and 14,440 bp in length. A total of 37 predicted genes were identified consisting of 13 protein-coding genes (PCGs), 22 transfer RNA (tRNAs) genes, two ribosomal RNA (rRNA) genes (rrnS and rrnL), and two non-coding regions. Interestingly, the assembled mitogenome has up to six times longer AT-rich regions than previous reference sequences, thus proving the advantage of long-read sequencing in resolving unreported non-coding regions. Furthermore, variant detection and phylogenetic analysis using concatenated protein coding genes, cox1, rrnL, and nd1 genes confirmed the distinct molecular identity of this newly assembled mitogenome while at the same time showing high genetic relationship with sequences from China or Tanzania. Our study provided a new set of reference mitogenome with better contiguity and resolved repetitive regions that could be used for meaningful phylogenetic analysis to further understand disease transmission and parasite biology.</p>\",\"PeriodicalId\":20260,\"journal\":{\"name\":\"PLoS Neglected Tropical Diseases\",\"volume\":\"17 8\",\"pages\":\"e0011586\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2023-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491297/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"PLoS Neglected Tropical Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1371/journal.pntd.0011586\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/8/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Neglected Tropical Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1371/journal.pntd.0011586","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
De novo assembled mitogenome analysis of Trichuris trichiura from Korean individuals using nanopore-based long-read sequencing technology.
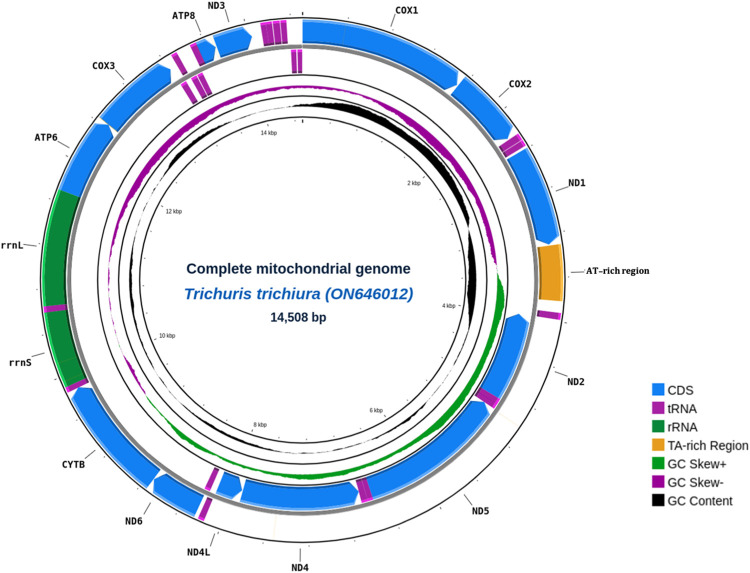
Knowledge about mitogenomes has been proven to be essential in human parasite diagnostics and understanding of their diversity. However, the lack of substantial data for comparative analysis is still a challenge in Trichuris trichiura research. To provide high quality mitogenomes, we utilized long-read sequencing technology of Oxford Nanopore Technologies (ONT) to better resolve repetitive regions and to construct de novo mitogenome assembly minimizing reference biases. In this study, we got three de novo assembled mitogenomes of T. trichiura isolated from Korean individuals. These circular complete mitogenomes of T. trichiura are 14,508 bp, 14,441 bp, and 14,440 bp in length. A total of 37 predicted genes were identified consisting of 13 protein-coding genes (PCGs), 22 transfer RNA (tRNAs) genes, two ribosomal RNA (rRNA) genes (rrnS and rrnL), and two non-coding regions. Interestingly, the assembled mitogenome has up to six times longer AT-rich regions than previous reference sequences, thus proving the advantage of long-read sequencing in resolving unreported non-coding regions. Furthermore, variant detection and phylogenetic analysis using concatenated protein coding genes, cox1, rrnL, and nd1 genes confirmed the distinct molecular identity of this newly assembled mitogenome while at the same time showing high genetic relationship with sequences from China or Tanzania. Our study provided a new set of reference mitogenome with better contiguity and resolved repetitive regions that could be used for meaningful phylogenetic analysis to further understand disease transmission and parasite biology.
期刊介绍:
PLOS Neglected Tropical Diseases publishes research devoted to the pathology, epidemiology, prevention, treatment and control of the neglected tropical diseases (NTDs), as well as relevant public policy.
The NTDs are defined as a group of poverty-promoting chronic infectious diseases, which primarily occur in rural areas and poor urban areas of low-income and middle-income countries. Their impact on child health and development, pregnancy, and worker productivity, as well as their stigmatizing features limit economic stability.
All aspects of these diseases are considered, including:
Pathogenesis
Clinical features
Pharmacology and treatment
Diagnosis
Epidemiology
Vector biology
Vaccinology and prevention
Demographic, ecological and social determinants
Public health and policy aspects (including cost-effectiveness analyses).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


