{"title":"用现代缩放算法纠正衍射数据中的系统误差。","authors":"Luis A Aldama, Kevin M Dalton, Doeke R Hekstra","doi":"10.1107/S2059798323005776","DOIUrl":null,"url":null,"abstract":"<p><p>X-ray diffraction enables the routine determination of the atomic structure of materials. Key to its success are data-processing algorithms that allow experimenters to determine the electron density of a sample from its diffraction pattern. Scaling, the estimation and correction of systematic errors in diffraction intensities, is an essential step in this process. These errors arise from sample heterogeneity, radiation damage, instrument limitations and other aspects of the experiment. New X-ray sources and sample-delivery methods, along with new experiments focused on changes in structure as a function of perturbations, have led to new demands on scaling algorithms. Classically, scaling algorithms use least-squares optimization to fit a model of common error sources to the observed diffraction intensities to force these intensities onto the same empirical scale. Recently, an alternative approach has been demonstrated which uses a Bayesian optimization method, variational inference, to simultaneously infer merged data along with corrections, or scale factors, for the systematic errors. Owing to its flexibility, this approach proves to be advantageous in certain scenarios. This perspective briefly reviews the history of scaling algorithms and contrasts them with variational inference. Finally, appropriate use cases are identified for the first such algorithm, Careless, guidance is offered on its use and some speculations are made about future variational scaling methods.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":"79 Pt 9","pages":"796-805"},"PeriodicalIF":2.6000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10478637/pdf/","citationCount":"0","resultStr":"{\"title\":\"Correcting systematic errors in diffraction data with modern scaling algorithms.\",\"authors\":\"Luis A Aldama, Kevin M Dalton, Doeke R Hekstra\",\"doi\":\"10.1107/S2059798323005776\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>X-ray diffraction enables the routine determination of the atomic structure of materials. Key to its success are data-processing algorithms that allow experimenters to determine the electron density of a sample from its diffraction pattern. Scaling, the estimation and correction of systematic errors in diffraction intensities, is an essential step in this process. These errors arise from sample heterogeneity, radiation damage, instrument limitations and other aspects of the experiment. New X-ray sources and sample-delivery methods, along with new experiments focused on changes in structure as a function of perturbations, have led to new demands on scaling algorithms. Classically, scaling algorithms use least-squares optimization to fit a model of common error sources to the observed diffraction intensities to force these intensities onto the same empirical scale. Recently, an alternative approach has been demonstrated which uses a Bayesian optimization method, variational inference, to simultaneously infer merged data along with corrections, or scale factors, for the systematic errors. Owing to its flexibility, this approach proves to be advantageous in certain scenarios. This perspective briefly reviews the history of scaling algorithms and contrasts them with variational inference. Finally, appropriate use cases are identified for the first such algorithm, Careless, guidance is offered on its use and some speculations are made about future variational scaling methods.</p>\",\"PeriodicalId\":7116,\"journal\":{\"name\":\"Acta Crystallographica. Section D, Structural Biology\",\"volume\":\"79 Pt 9\",\"pages\":\"796-805\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2023-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10478637/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Crystallographica. Section D, Structural Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1107/S2059798323005776\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/8/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798323005776","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
X 射线衍射技术可对材料的原子结构进行常规测定。其成功的关键在于数据处理算法,实验人员可以通过衍射图样确定样品的电子密度。缩放,即估计和修正衍射强度的系统误差,是这一过程中必不可少的一步。这些误差来自样品的异质性、辐射损伤、仪器限制和实验的其他方面。新的 X 射线源和样品输送方法,以及侧重于结构变化作为扰动函数的新实验,都对缩放算法提出了新的要求。经典的缩放算法使用最小二乘优化方法,将常见误差源模型拟合到观测到的衍射强度上,从而迫使这些强度达到相同的经验尺度。最近,人们展示了另一种方法,即使用贝叶斯优化方法(变异推理),同时推断合并数据以及系统误差的修正或比例因子。由于其灵活性,这种方法在某些情况下证明是有优势的。本视角简要回顾了缩放算法的历史,并将其与变分推理进行了对比。最后,确定了第一种此类算法 Careless 的适当用例,为其使用提供了指导,并对未来的变分缩放方法做了一些推测。
Correcting systematic errors in diffraction data with modern scaling algorithms.
X-ray diffraction enables the routine determination of the atomic structure of materials. Key to its success are data-processing algorithms that allow experimenters to determine the electron density of a sample from its diffraction pattern. Scaling, the estimation and correction of systematic errors in diffraction intensities, is an essential step in this process. These errors arise from sample heterogeneity, radiation damage, instrument limitations and other aspects of the experiment. New X-ray sources and sample-delivery methods, along with new experiments focused on changes in structure as a function of perturbations, have led to new demands on scaling algorithms. Classically, scaling algorithms use least-squares optimization to fit a model of common error sources to the observed diffraction intensities to force these intensities onto the same empirical scale. Recently, an alternative approach has been demonstrated which uses a Bayesian optimization method, variational inference, to simultaneously infer merged data along with corrections, or scale factors, for the systematic errors. Owing to its flexibility, this approach proves to be advantageous in certain scenarios. This perspective briefly reviews the history of scaling algorithms and contrasts them with variational inference. Finally, appropriate use cases are identified for the first such algorithm, Careless, guidance is offered on its use and some speculations are made about future variational scaling methods.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.

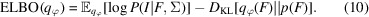


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


