{"title":"通过与单个样品的比较,评估Infinium MethylationEPIC BeadChip阵列的混合样品方法。","authors":"Shota Nishitani, Takashi X Fujisawa, Akiko Yao, Shinichiro Takiguchi, Akemi Tomoda","doi":"10.1186/s13148-023-01544-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The pooled sample method is used in epigenomic research and expression analysis and is a cost-effective screening approach for small amounts of DNA. Evaluation of the pooled sample method in epigenomic studies is performed using the Illumina Infinium Methylation 450K BeadChip array; however, subsequent reports on the updated 850K array are lacking. A previous study demonstrated that the methylation levels obtained from individual samples were accurately replicated using pooled samples but did not address epigenome-wide association study (EWAS) statistics. The DNA quantification method, which is important for the homogeneous mixing of DNA in the pooled sample method, has since become fluorescence-based, and additional factors need to be considered including the resolution of batch effects of microarray chips and the heterogeneity of the cellular proportions from which the DNA samples are derived. In this study, four pooled samples were created from 44 individual samples, and EWAS statistics for differentially methylated positions (DMPs) and regions (DMRs) were conducted for individual samples and compared with the statistics obtained from the pooled samples.</p><p><strong>Results: </strong>The methylation levels could be reproduced fairly well in the pooled samples. This was the case for the entire dataset and when limited to the top 100 CpG sites, consistent with a previous study using the 450K BeadChip array. However, the statistical results of the EWAS for the DMP by individual samples were not replicated in pooled samples. Qualitative analyses highlighting methylation within an arbitrary candidate gene were replicable. Focusing on chr 20, the statistical results of EWAS for DMR from individual samples showed replicability in the pooled samples as long as they were limited to regions with a sufficient effect size.</p><p><strong>Conclusions: </strong>The pooled sample method replicated the methylation values well and can be used for EWAS in DMR. This method is sample amount-effective and cost-effective and can be utilized for screening by carefully understanding the effective features and disadvantages of the pooled sample method and combining it with candidate gene analyses.</p>","PeriodicalId":48652,"journal":{"name":"Clinical Epigenetics","volume":"15 1","pages":"138"},"PeriodicalIF":4.4000,"publicationDate":"2023-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10463626/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evaluation of the pooled sample method in Infinium MethylationEPIC BeadChip array by comparison with individual samples.\",\"authors\":\"Shota Nishitani, Takashi X Fujisawa, Akiko Yao, Shinichiro Takiguchi, Akemi Tomoda\",\"doi\":\"10.1186/s13148-023-01544-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The pooled sample method is used in epigenomic research and expression analysis and is a cost-effective screening approach for small amounts of DNA. Evaluation of the pooled sample method in epigenomic studies is performed using the Illumina Infinium Methylation 450K BeadChip array; however, subsequent reports on the updated 850K array are lacking. A previous study demonstrated that the methylation levels obtained from individual samples were accurately replicated using pooled samples but did not address epigenome-wide association study (EWAS) statistics. The DNA quantification method, which is important for the homogeneous mixing of DNA in the pooled sample method, has since become fluorescence-based, and additional factors need to be considered including the resolution of batch effects of microarray chips and the heterogeneity of the cellular proportions from which the DNA samples are derived. In this study, four pooled samples were created from 44 individual samples, and EWAS statistics for differentially methylated positions (DMPs) and regions (DMRs) were conducted for individual samples and compared with the statistics obtained from the pooled samples.</p><p><strong>Results: </strong>The methylation levels could be reproduced fairly well in the pooled samples. This was the case for the entire dataset and when limited to the top 100 CpG sites, consistent with a previous study using the 450K BeadChip array. However, the statistical results of the EWAS for the DMP by individual samples were not replicated in pooled samples. Qualitative analyses highlighting methylation within an arbitrary candidate gene were replicable. Focusing on chr 20, the statistical results of EWAS for DMR from individual samples showed replicability in the pooled samples as long as they were limited to regions with a sufficient effect size.</p><p><strong>Conclusions: </strong>The pooled sample method replicated the methylation values well and can be used for EWAS in DMR. This method is sample amount-effective and cost-effective and can be utilized for screening by carefully understanding the effective features and disadvantages of the pooled sample method and combining it with candidate gene analyses.</p>\",\"PeriodicalId\":48652,\"journal\":{\"name\":\"Clinical Epigenetics\",\"volume\":\"15 1\",\"pages\":\"138\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2023-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10463626/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Epigenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13148-023-01544-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-023-01544-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
Evaluation of the pooled sample method in Infinium MethylationEPIC BeadChip array by comparison with individual samples.
Background: The pooled sample method is used in epigenomic research and expression analysis and is a cost-effective screening approach for small amounts of DNA. Evaluation of the pooled sample method in epigenomic studies is performed using the Illumina Infinium Methylation 450K BeadChip array; however, subsequent reports on the updated 850K array are lacking. A previous study demonstrated that the methylation levels obtained from individual samples were accurately replicated using pooled samples but did not address epigenome-wide association study (EWAS) statistics. The DNA quantification method, which is important for the homogeneous mixing of DNA in the pooled sample method, has since become fluorescence-based, and additional factors need to be considered including the resolution of batch effects of microarray chips and the heterogeneity of the cellular proportions from which the DNA samples are derived. In this study, four pooled samples were created from 44 individual samples, and EWAS statistics for differentially methylated positions (DMPs) and regions (DMRs) were conducted for individual samples and compared with the statistics obtained from the pooled samples.
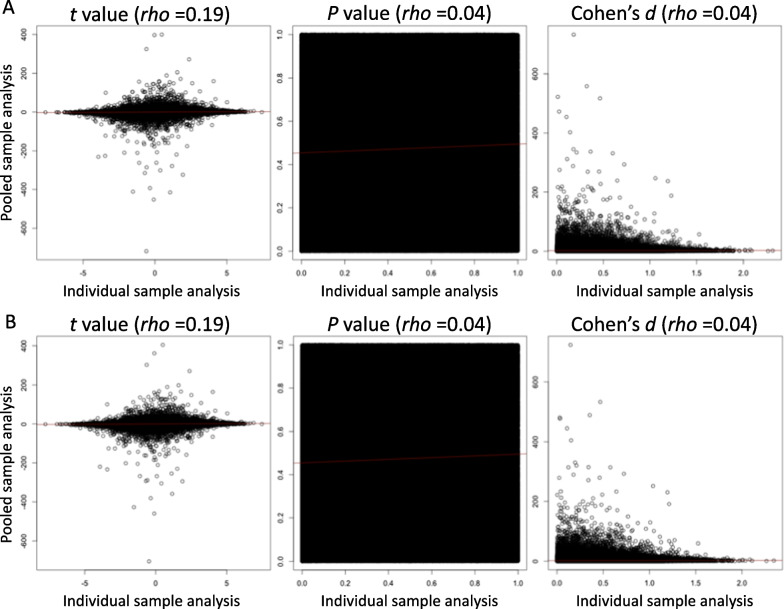
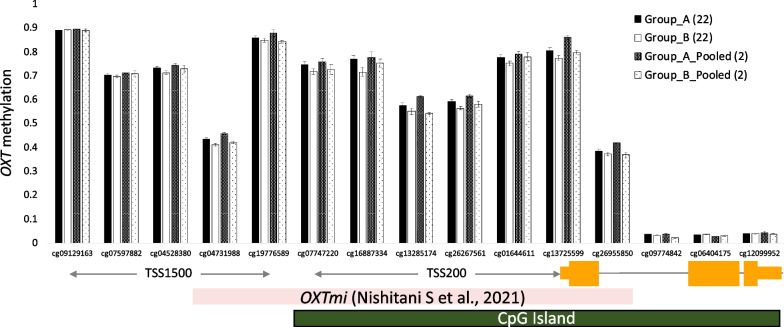
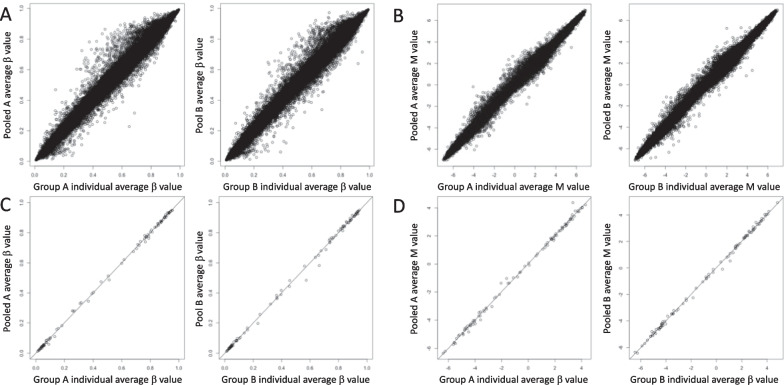
Results: The methylation levels could be reproduced fairly well in the pooled samples. This was the case for the entire dataset and when limited to the top 100 CpG sites, consistent with a previous study using the 450K BeadChip array. However, the statistical results of the EWAS for the DMP by individual samples were not replicated in pooled samples. Qualitative analyses highlighting methylation within an arbitrary candidate gene were replicable. Focusing on chr 20, the statistical results of EWAS for DMR from individual samples showed replicability in the pooled samples as long as they were limited to regions with a sufficient effect size.
Conclusions: The pooled sample method replicated the methylation values well and can be used for EWAS in DMR. This method is sample amount-effective and cost-effective and can be utilized for screening by carefully understanding the effective features and disadvantages of the pooled sample method and combining it with candidate gene analyses.
Clinical EpigeneticsBiochemistry, Genetics and Molecular Biology-Developmental Biology
CiteScore
8.90
自引率
5.30%
发文量
150
审稿时长
12 weeks
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


