Jun Young Park, Jang Jae Lee, Younghwa Lee, Dongsoo Lee, Jungsoo Gim, Lindsay Farrer, Kun Ho Lee, Sungho Won
{"title":"基于机器学习的疾病不确定性量化增加了遗传关联研究的统计能力。","authors":"Jun Young Park, Jang Jae Lee, Younghwa Lee, Dongsoo Lee, Jungsoo Gim, Lindsay Farrer, Kun Ho Lee, Sungho Won","doi":"10.1093/bioinformatics/btad534","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Allowance for increasingly large samples is a key to identify the association of genetic variants with Alzheimer's disease (AD) in genome-wide association studies (GWAS). Accordingly, we aimed to develop a method that incorporates patients with mild cognitive impairment and unknown cognitive status in GWAS using a machine learning-based AD prediction model.</p><p><strong>Results: </strong>Simulation analyses showed that weighting imputed phenotypes method increased the statistical power compared to ordinary logistic regression using only AD cases and controls. Applied to real-world data, the penalized logistic method had the highest AUC (0.96) for AD prediction and weighting imputed phenotypes method performed well in terms of power. We identified an association (P<5.0×10-8) of AD with several variants in the APOE region and rs143625563 in LMX1A. Our method, which allows the inclusion of individuals with mild cognitive impairment, improves the statistical power of GWAS for AD. We discovered a novel association with LMX1A.</p><p><strong>Availability and implementation: </strong>Simulation codes can be accessed at https://github.com/Junkkkk/wGEE_GWAS.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10539075/pdf/","citationCount":"0","resultStr":"{\"title\":\"Machine learning-based quantification for disease uncertainty increases the statistical power of genetic association studies.\",\"authors\":\"Jun Young Park, Jang Jae Lee, Younghwa Lee, Dongsoo Lee, Jungsoo Gim, Lindsay Farrer, Kun Ho Lee, Sungho Won\",\"doi\":\"10.1093/bioinformatics/btad534\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Allowance for increasingly large samples is a key to identify the association of genetic variants with Alzheimer's disease (AD) in genome-wide association studies (GWAS). Accordingly, we aimed to develop a method that incorporates patients with mild cognitive impairment and unknown cognitive status in GWAS using a machine learning-based AD prediction model.</p><p><strong>Results: </strong>Simulation analyses showed that weighting imputed phenotypes method increased the statistical power compared to ordinary logistic regression using only AD cases and controls. Applied to real-world data, the penalized logistic method had the highest AUC (0.96) for AD prediction and weighting imputed phenotypes method performed well in terms of power. We identified an association (P<5.0×10-8) of AD with several variants in the APOE region and rs143625563 in LMX1A. Our method, which allows the inclusion of individuals with mild cognitive impairment, improves the statistical power of GWAS for AD. We discovered a novel association with LMX1A.</p><p><strong>Availability and implementation: </strong>Simulation codes can be accessed at https://github.com/Junkkkk/wGEE_GWAS.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10539075/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad534\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad534","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Machine learning-based quantification for disease uncertainty increases the statistical power of genetic association studies.
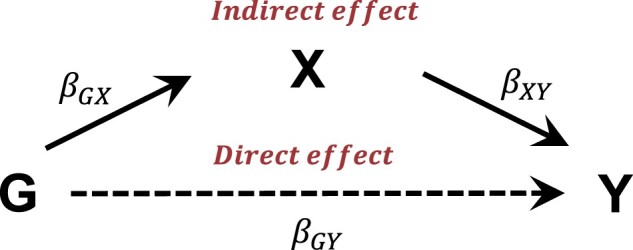
Motivation: Allowance for increasingly large samples is a key to identify the association of genetic variants with Alzheimer's disease (AD) in genome-wide association studies (GWAS). Accordingly, we aimed to develop a method that incorporates patients with mild cognitive impairment and unknown cognitive status in GWAS using a machine learning-based AD prediction model.
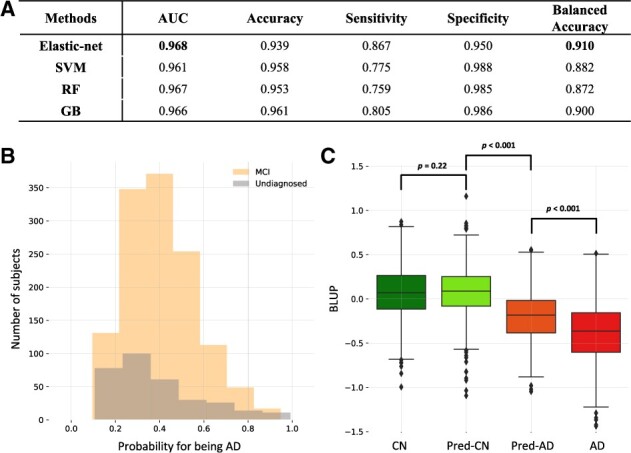
Results: Simulation analyses showed that weighting imputed phenotypes method increased the statistical power compared to ordinary logistic regression using only AD cases and controls. Applied to real-world data, the penalized logistic method had the highest AUC (0.96) for AD prediction and weighting imputed phenotypes method performed well in terms of power. We identified an association (P<5.0×10-8) of AD with several variants in the APOE region and rs143625563 in LMX1A. Our method, which allows the inclusion of individuals with mild cognitive impairment, improves the statistical power of GWAS for AD. We discovered a novel association with LMX1A.
Availability and implementation: Simulation codes can be accessed at https://github.com/Junkkkk/wGEE_GWAS.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


