Spontaneous allelic variant in deafness–blindness gene Ush1g resulting in an expanded phenotype
IF 2.3
4区 心理学
Q2 BEHAVIORAL SCIENCES
引用次数: 0
Abstract
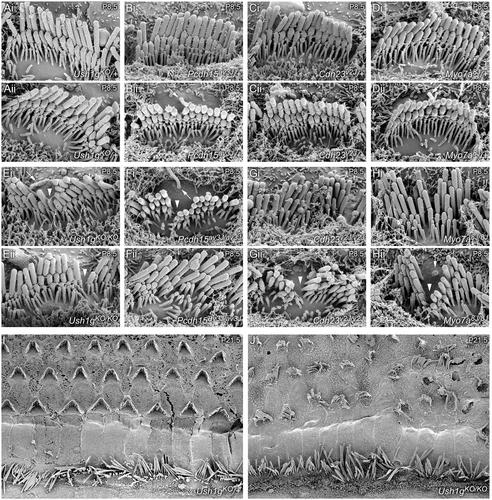
Relationships between novel phenotypic behaviors and specific genetic alterations are often discovered using target‐specific, directed mutagenesis or phenotypic selection following chemical mutagenesis. An alternative approach is to exploit deficiencies in DNA repair pathways that maintain genetic integrity in response to spontaneously induced damage. Mice deficient in the DNA glycosylase NEIL1 show elevated spontaneous mutations, which arise from translesion DNA synthesis past oxidatively induced base damage. Several litters of Neil1 knockout mice included animals that were distinguished by their backwards‐walking behavior in open‐field environments, while maintaining frantic forward movements in their home cage environment. Other phenotypic manifestations included swim test failures, head tilting and circling. Mapping of the mutation that conferred these behaviors showed the introduction of a stop codon at amino acid 4 of the Ush1g gene. Ush1gbw/bw null mice displayed auditory and vestibular defects that are commonly seen with mutations affecting inner‐ear hair‐cell function, including a complete lack of auditory brainstem responses and vestibular‐evoked potentials. As in other Usher syndrome type I mutant mouse lines, hair cell phenotypes included disorganized and split hair bundles, as well as altered distribution of proteins for stereocilia that localize to the tips of row 1 or row 2. Disruption to the bundle and kinocilium displacement suggested that USH1G is essential for forming the hair cell's kinocilial links. Consistent with other Usher type 1 models, Ush1gbw/bw mice had no substantial retinal degeneration compared with Ush1gbw/+ controls. In contrast to previously described Ush1g alleles, this new allele provides the first knockout model for this gene.
聋盲基因Ush1g的自发等位变异导致表型扩大
新的表型行为和特定遗传改变之间的关系通常是通过靶向性、定向诱变或化学诱变后的表型选择来发现的。另一种方法是利用DNA修复途径中的缺陷来维持遗传完整性,以应对自发诱导的损伤。DNA糖基酶NEIL1缺乏的小鼠表现出较高的自发突变,这是由于翻译DNA合成经过氧化诱导的碱基损伤引起的。在几窝Neil1基因敲除小鼠中,有一些动物在开阔环境中表现出向后行走的行为,而在笼子环境中则保持疯狂的向前运动。其他表型表现包括游泳试验失败,头部倾斜和盘旋。赋予这些行为的突变图谱显示,在Ush1g基因的氨基酸4处引入了一个停止密码子。Ush1gbw/bw小鼠表现出听觉和前庭功能缺陷,这些缺陷在影响内耳毛细胞功能的突变中很常见,包括完全缺乏听觉脑干反应和前庭诱发电位。与其他Usher综合征I型突变小鼠系一样,毛细胞表型包括无组织和分裂的毛束,以及位于第1行或第2行尖端的立纤毛蛋白质分布的改变。束的破坏和运动纤毛移位表明USH1G对毛细胞运动纤毛连接的形成至关重要。与其他Usher 1型模型一致,与Ush1gbw/+对照组相比,Ush1gbw/bw小鼠没有明显的视网膜变性。与先前描述的Ush1g等位基因相比,这个新的等位基因提供了该基因的第一个敲除模型。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Genes Brain and Behavior
医学-行为科学
CiteScore
6.80
自引率
4.00%
发文量
62
审稿时长
4-8 weeks
期刊介绍:
Genes, Brain and Behavior was launched in 2002 with the aim of publishing top quality research in behavioral and neural genetics in their broadest sense. The emphasis is on the analysis of the behavioral and neural phenotypes under consideration, the unifying theme being the genetic approach as a tool to increase our understanding of these phenotypes.
Genes Brain and Behavior is pleased to offer the following features:
8 issues per year
online submissions with first editorial decisions within 3-4 weeks and fast publication at Wiley-Blackwells
High visibility through its coverage by PubMed/Medline, Current Contents and other major abstracting and indexing services
Inclusion in the Wiley-Blackwell consortial license, extending readership to thousands of international libraries and institutions
A large and varied editorial board comprising of international specialists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


