{"title":"Clinical, Laboratory, Radiological, and Genetic Characteristics of Pediatric Patients with Alagille Syndrome.","authors":"Hasan M Isa, Fawzeya A Alahmed","doi":"10.4103/abr.abr_201_22","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Alagille syndrome (ALGS) is an autosomal dominant disease caused by <i>JAG1</i> or <i>NOTCH2</i> mutation. It is diagnosed by the presence of three out of five features: characteristic facies, posterior embryotoxon, peripheral pulmonary stenosis, vertebral defects, and interlobular bile duct paucity. This study aimed to review the prevalence, clinical presentations, diagnosis, treatment, and outcome of patients with ALGS.</p><p><strong>Materials and methods: </strong>This is a retrospective review of patients with ALGS at the Pediatric Department, Salmaniya Medical Complex, Bahrain, between August 1994 and October 2022. The diagnosis was based on clinical, laboratory, radiological, histopathological, and genetic findings.</p><p><strong>Results: </strong>Five patients were found to have ALGS. The prevalence of ALGS in Bahrain was 1.04 patients per 100,000 (0.001%). Four were Bahraini and three were females. Median birth weight was 2.3 (2.3-2.5) kg. All patients presented at the time of birth with low birth weight, cholestatic jaundice, clay-colored stool, heart murmur, and dysmorphic facial features. All had congenital heart diseases, two had butterfly vertebrae, and one had posterior embryotoxon. All had elevated liver enzymes and normal abdominal ultrasound. Three had positive hepatobiliary iminodiacetic acid scan and one had bile duct paucity in liver biopsy. Three had intraoperative cholangiogram. Four were positive for <i>JAG1</i> mutation. All received ursodeoxycholic acid and fat-soluble vitamins. Two required liver transplantation.</p><p><strong>Conclusion: </strong>ALGS is a rare disorder in Bahrain. Diagnosis is challenging as the disease can be associated with or misdiagnosed as biliary atresia. Patients with ALGS are at high risk of morbidity either by unnecessary intraoperative cholangiogram or unavoidable liver transplantation.</p>","PeriodicalId":7225,"journal":{"name":"Advanced Biomedical Research","volume":"12 ","pages":"155"},"PeriodicalIF":0.0000,"publicationDate":"2023-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/71/91/ABR-12-155.PMC10410416.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Biomedical Research","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4103/abr.abr_201_22","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Alagille syndrome (ALGS) is an autosomal dominant disease caused by JAG1 or NOTCH2 mutation. It is diagnosed by the presence of three out of five features: characteristic facies, posterior embryotoxon, peripheral pulmonary stenosis, vertebral defects, and interlobular bile duct paucity. This study aimed to review the prevalence, clinical presentations, diagnosis, treatment, and outcome of patients with ALGS.
Materials and methods: This is a retrospective review of patients with ALGS at the Pediatric Department, Salmaniya Medical Complex, Bahrain, between August 1994 and October 2022. The diagnosis was based on clinical, laboratory, radiological, histopathological, and genetic findings.
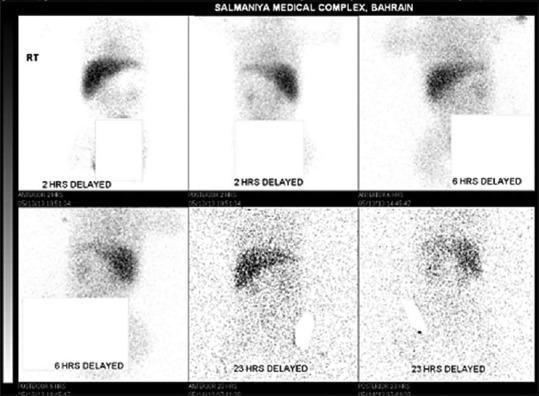
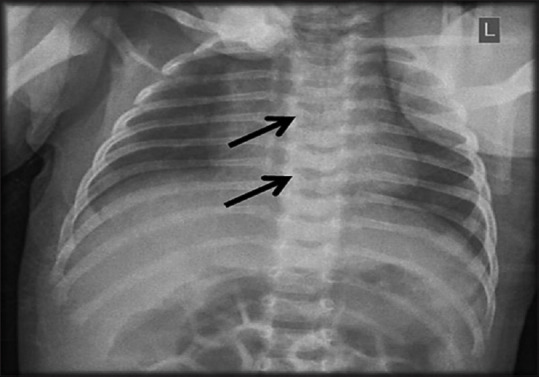
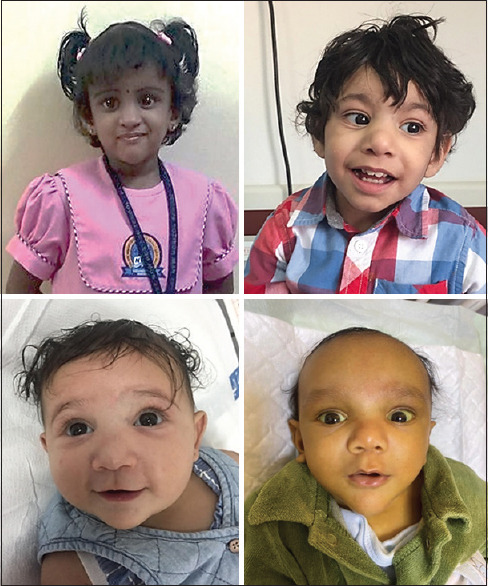
Results: Five patients were found to have ALGS. The prevalence of ALGS in Bahrain was 1.04 patients per 100,000 (0.001%). Four were Bahraini and three were females. Median birth weight was 2.3 (2.3-2.5) kg. All patients presented at the time of birth with low birth weight, cholestatic jaundice, clay-colored stool, heart murmur, and dysmorphic facial features. All had congenital heart diseases, two had butterfly vertebrae, and one had posterior embryotoxon. All had elevated liver enzymes and normal abdominal ultrasound. Three had positive hepatobiliary iminodiacetic acid scan and one had bile duct paucity in liver biopsy. Three had intraoperative cholangiogram. Four were positive for JAG1 mutation. All received ursodeoxycholic acid and fat-soluble vitamins. Two required liver transplantation.
Conclusion: ALGS is a rare disorder in Bahrain. Diagnosis is challenging as the disease can be associated with or misdiagnosed as biliary atresia. Patients with ALGS are at high risk of morbidity either by unnecessary intraoperative cholangiogram or unavoidable liver transplantation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


