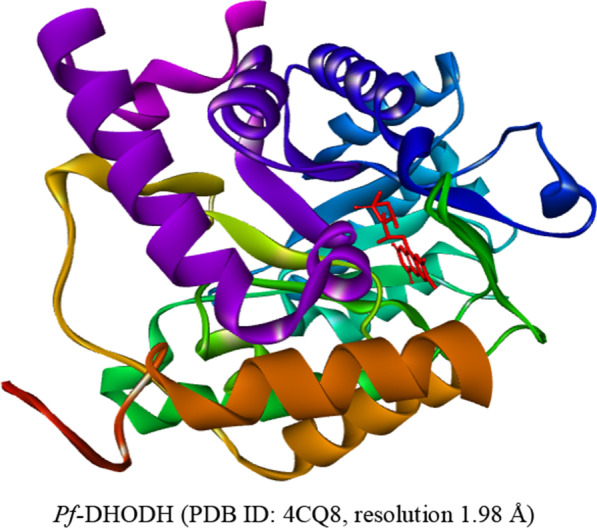
Virtual screening and molecular dynamic simulations of the antimalarial derivatives of 2-anilino 4-amino substituted quinazolines docked against a Pf-DHODH protein target.
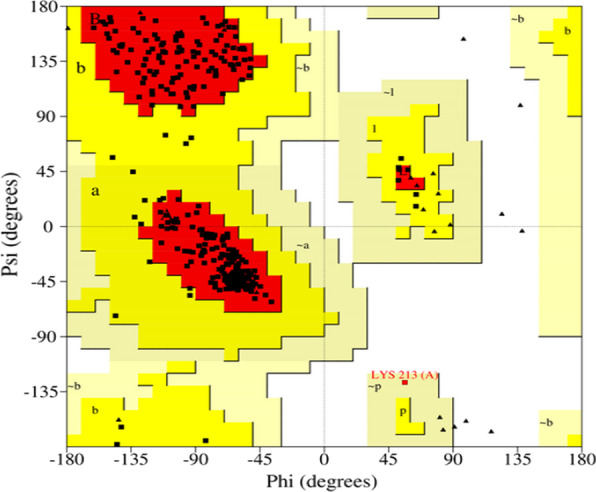
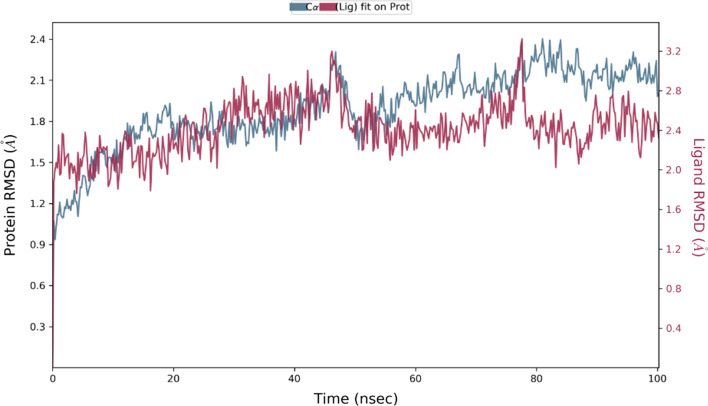
{"title":"Virtual screening and molecular dynamic simulations of the antimalarial derivatives of 2-anilino 4-amino substituted quinazolines docked against a <i>Pf</i>-DHODH protein target.","authors":"Zakari Ya'u Ibrahim, Adamu Uzairu, Gideon Adamu Shallangwa, Stephen Eyije Abechi, Sulaiman Isyaku","doi":"10.1186/s43042-022-00329-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The processes of drug development and validation are too expensive to be subjected to experimental trial and errors. Hence, the use of the insilico approach becomes imperative. To this effect, the drug-likeness and pharmacokinetic properties of the ten (10) previously designed derivatives of 2-anilino 4-amino substituted quinazolines were carried out. Their predicted ligand binding interactions were also carried out by docking them against the <i>Plasmodium falciparum</i> dihydroorotate dehydrogenase (<i>Pf-</i>DHODH) protein target, and the stability of the complex was determined through dynamic simulations. The drug-likeness and pharmacokinetic characteristics were estimated using the online SwissADME software, while the Molegro Virtual Docker (MVD) software was used for molecular docking. And the dynamic simulation was performed for the duration of 100 ns to verify the stability of the docked complex, with the aid of a Schrödinger program, Desmond.</p><p><strong>Results: </strong>The designed derivatives were all found to pass the Lipinski test of drug likeness, while the pharmacokinetic studies result that the skin permeability and molar refractivity values of the derivatives are both within the limits. In addition, except for derivative C-01, most of the derivatives have strong gastrointestinal absorptions and lack Pgp substrate. Furthermore, no derivative inhibited CYP1A2, CYP2C9, or CYP2C19. The docking studies show the better binding affinities between the ligands and <i>Pf-</i>DHODH than those between the atovaquone or chloroquine standards. The derivative C-02, {5-((6,7-dimethoxy-4-((3-nitrobenzyl)amino)quinazolin-2-yl)amino)-2-fluorobenzaldehyde} was found to be the most stable derivative, with a re-rank docking score of - 173.528 kcal/mol and interaction energy of - 225.112 kcal/mol. The dynamic simulation analysis shows that the derivative C-02 forms a stable complex with the protein target over the simulation time.</p><p><strong>Conclusions: </strong>The ability of these ligands to form hydrogen bonds, as well as various other interactions, was cited as a factor responsible for their better binding affinity. These findings could aid further the development of enhanced antimalarial drugs.</p>","PeriodicalId":74994,"journal":{"name":"The Egyptian journal of medical human genetics","volume":"23 1","pages":"119"},"PeriodicalIF":1.1000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9364290/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Egyptian journal of medical human genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s43042-022-00329-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/8/10 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The processes of drug development and validation are too expensive to be subjected to experimental trial and errors. Hence, the use of the insilico approach becomes imperative. To this effect, the drug-likeness and pharmacokinetic properties of the ten (10) previously designed derivatives of 2-anilino 4-amino substituted quinazolines were carried out. Their predicted ligand binding interactions were also carried out by docking them against the Plasmodium falciparum dihydroorotate dehydrogenase (Pf-DHODH) protein target, and the stability of the complex was determined through dynamic simulations. The drug-likeness and pharmacokinetic characteristics were estimated using the online SwissADME software, while the Molegro Virtual Docker (MVD) software was used for molecular docking. And the dynamic simulation was performed for the duration of 100 ns to verify the stability of the docked complex, with the aid of a Schrödinger program, Desmond.
Results: The designed derivatives were all found to pass the Lipinski test of drug likeness, while the pharmacokinetic studies result that the skin permeability and molar refractivity values of the derivatives are both within the limits. In addition, except for derivative C-01, most of the derivatives have strong gastrointestinal absorptions and lack Pgp substrate. Furthermore, no derivative inhibited CYP1A2, CYP2C9, or CYP2C19. The docking studies show the better binding affinities between the ligands and Pf-DHODH than those between the atovaquone or chloroquine standards. The derivative C-02, {5-((6,7-dimethoxy-4-((3-nitrobenzyl)amino)quinazolin-2-yl)amino)-2-fluorobenzaldehyde} was found to be the most stable derivative, with a re-rank docking score of - 173.528 kcal/mol and interaction energy of - 225.112 kcal/mol. The dynamic simulation analysis shows that the derivative C-02 forms a stable complex with the protein target over the simulation time.
Conclusions: The ability of these ligands to form hydrogen bonds, as well as various other interactions, was cited as a factor responsible for their better binding affinity. These findings could aid further the development of enhanced antimalarial drugs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


