Waqas Jehangir, Alexander D Karabachev, Elvira R Umyarova
{"title":"Pulmonary Haemosiderosis Secondary to Hereditary Haemochromatosis; a Case Report.","authors":"Waqas Jehangir, Alexander D Karabachev, Elvira R Umyarova","doi":"10.37029/jcas.v6i1.281","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Hereditary haemochromatosis (HH) is an autosomal recessive disease of increased intestinal absorption of iron, leading to accumulation in tissues which may progress to organ damage, most commonly in the liver. Iron deposition in the liver can lead to cirrhosis and hepatocellular carcinoma. Other common manifestations of haemochromatosis include diabetes, bronzing of the skin, arthropathy and cardiomyopathy. Here, we describe a case of pulmonary haemosiderosis secondary to HH.</p><p><strong>Case description: </strong>A 49-year-old male with no medical history or family history of iron overload presented with fatigue, shortness of breath and chest pain after a recent finding of elevated ferritin. The patient was found to have biallelic C282Y mutations of the human homeostatic iron regulator protein (<i>HFE</i>) protein and after further workup with laboratory tests and imaging was diagnosed with HH with secondary pulmonary haemosiderosis. The patient is receiving twice weekly phlebotomies and has had an overall improvement in his symptoms.</p><p><strong>Practical implications: </strong>The presentation of haemochromatosis can vary widely depending on the severity of iron overload and the presence of conditions that predispose organ dysfunction. Pulmonary haemosiderosis is a very rare manifestation of HH. This report illustrates the various manifestations of this disease and provides insight into this rare presentation to improve the diagnosis of this disease.</p>","PeriodicalId":73631,"journal":{"name":"Journal of cancer & allied specialties","volume":"6 1","pages":"e281"},"PeriodicalIF":0.0000,"publicationDate":"2020-01-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/68/c4/JCAS-6-281.PMC10166317.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of cancer & allied specialties","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.37029/jcas.v6i1.281","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
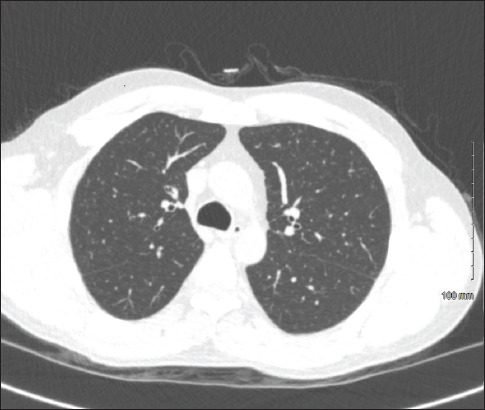
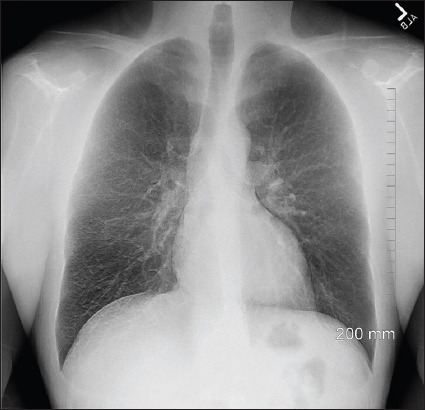
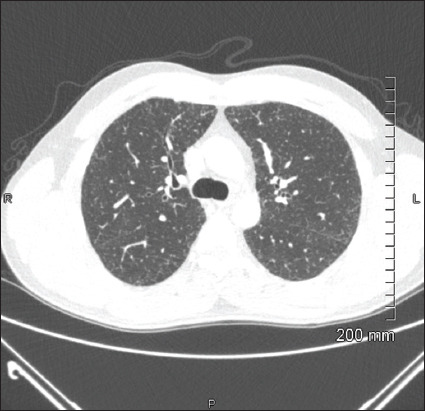
Introduction: Hereditary haemochromatosis (HH) is an autosomal recessive disease of increased intestinal absorption of iron, leading to accumulation in tissues which may progress to organ damage, most commonly in the liver. Iron deposition in the liver can lead to cirrhosis and hepatocellular carcinoma. Other common manifestations of haemochromatosis include diabetes, bronzing of the skin, arthropathy and cardiomyopathy. Here, we describe a case of pulmonary haemosiderosis secondary to HH.
Case description: A 49-year-old male with no medical history or family history of iron overload presented with fatigue, shortness of breath and chest pain after a recent finding of elevated ferritin. The patient was found to have biallelic C282Y mutations of the human homeostatic iron regulator protein (HFE) protein and after further workup with laboratory tests and imaging was diagnosed with HH with secondary pulmonary haemosiderosis. The patient is receiving twice weekly phlebotomies and has had an overall improvement in his symptoms.
Practical implications: The presentation of haemochromatosis can vary widely depending on the severity of iron overload and the presence of conditions that predispose organ dysfunction. Pulmonary haemosiderosis is a very rare manifestation of HH. This report illustrates the various manifestations of this disease and provides insight into this rare presentation to improve the diagnosis of this disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


