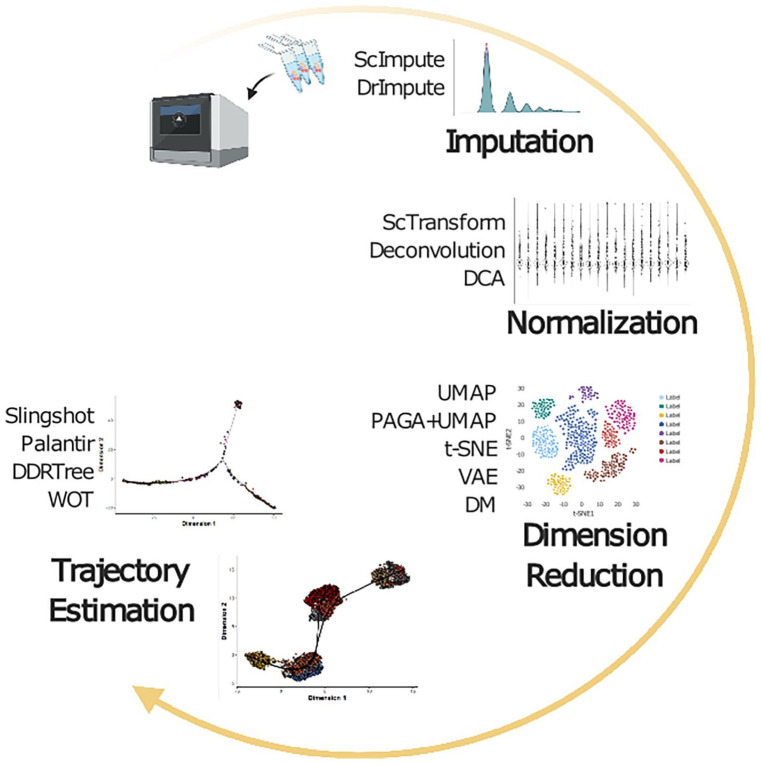
{"title":"Stability of scRNA-Seq Analysis Workflows is Susceptible to Preprocessing and is Mitigated by Regularized or Supervised Approaches.","authors":"Arda Durmaz, Jacob G Scott","doi":"10.1177/11769343221123050","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Statistical methods developed to address various questions in single-cell datasets show increased variability to different parameter regimes. In order to delineate further the robustness of commonly utilized methods for single-cell RNA-Seq, we aimed to comprehensively review scRNA-Seq analysis workflows in the setting of dimension reduction, clustering, and trajectory inference.</p><p><strong>Methods: </strong>We utilized datasets with temporal single-cell transcriptomics profiles from public repositories. Combining multiple methods at each level of the workflow, we have performed over 6<i>k</i> analysis and evaluated the results of clustering and pseudotime estimation using adjusted rand index and rank correlation metrics. We have further integrated neural network methods to assess whether models with increased complexity can show increased bias/variance trade-off.</p><p><strong>Results: </strong>Combinatorial workflows showed that utilizing non-linear dimension reduction techniques such as t-SNE and UMAP are sensitive to initial preprocessing steps hence clustering results on dimension reduced space of single-cell datasets should be utilized carefully. Similarly, pseudotime estimation methods that depend on previous non-linear dimension reduction steps can result in highly variable trajectories. In contrast, methods that avoid non-linearity such as WOT can result in repeatable inferences of temporal gene expression dynamics. Furthermore, imputation methods do not improve clustering or trajectory inference results substantially in terms of repeatability. In contrast, the selection of the normalization method shows an increased effect on downstream analysis where ScTransform reduces variability overall.</p>","PeriodicalId":50472,"journal":{"name":"Evolutionary Bioinformatics","volume":"18 ","pages":"11769343221123050"},"PeriodicalIF":1.7000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/07/96/10.1177_11769343221123050.PMC9527995.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/11769343221123050","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
Background: Statistical methods developed to address various questions in single-cell datasets show increased variability to different parameter regimes. In order to delineate further the robustness of commonly utilized methods for single-cell RNA-Seq, we aimed to comprehensively review scRNA-Seq analysis workflows in the setting of dimension reduction, clustering, and trajectory inference.
Methods: We utilized datasets with temporal single-cell transcriptomics profiles from public repositories. Combining multiple methods at each level of the workflow, we have performed over 6k analysis and evaluated the results of clustering and pseudotime estimation using adjusted rand index and rank correlation metrics. We have further integrated neural network methods to assess whether models with increased complexity can show increased bias/variance trade-off.
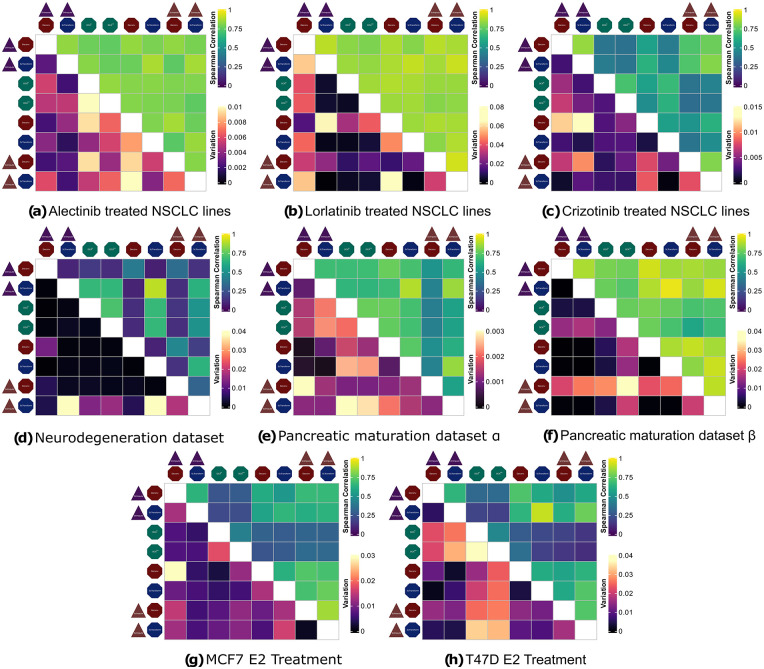
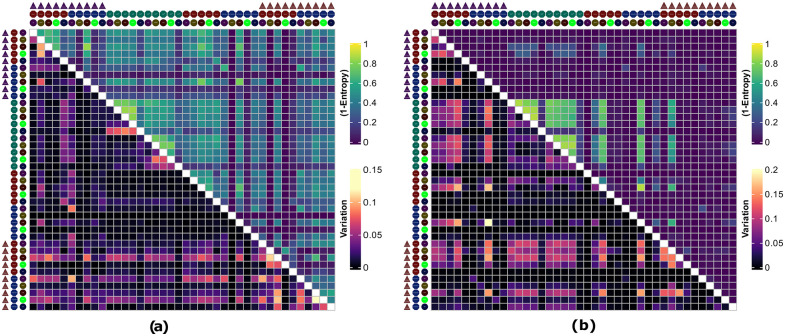
Results: Combinatorial workflows showed that utilizing non-linear dimension reduction techniques such as t-SNE and UMAP are sensitive to initial preprocessing steps hence clustering results on dimension reduced space of single-cell datasets should be utilized carefully. Similarly, pseudotime estimation methods that depend on previous non-linear dimension reduction steps can result in highly variable trajectories. In contrast, methods that avoid non-linearity such as WOT can result in repeatable inferences of temporal gene expression dynamics. Furthermore, imputation methods do not improve clustering or trajectory inference results substantially in terms of repeatability. In contrast, the selection of the normalization method shows an increased effect on downstream analysis where ScTransform reduces variability overall.
期刊介绍:
Evolutionary Bioinformatics is an open access, peer reviewed international journal focusing on evolutionary bioinformatics. The journal aims to support understanding of organismal form and function through use of molecular, genetic, genomic and proteomic data by giving due consideration to its evolutionary context.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


