Charles Spanbauer, Wei Pan, ADNI, The Alzheimer's Disease Neuroimaging Initiative
{"title":"Sparse prediction informed by genetic annotations using the logit normal prior for Bayesian regression tree ensembles","authors":"Charles Spanbauer, Wei Pan, ADNI, The Alzheimer's Disease Neuroimaging Initiative","doi":"10.1002/gepi.22505","DOIUrl":null,"url":null,"abstract":"<p>Using high-dimensional genetic variants such as single nucleotide polymorphisms (SNP) to predict complex diseases and traits has important applications in basic research and other clinical settings. For example, predicting gene expression is a necessary first step to identify (putative) causal genes in transcriptome-wide association studies. Due to weak signals, high-dimensionality, and linkage disequilibrium (correlation) among SNPs, building such a prediction model is challenging. However, functional annotations at the SNP level (e.g., as epigenomic data across multiple cell- or tissue-types) are available and could be used to inform predictor importance and aid in outcome prediction. Existing approaches to incorporate annotations have been based mainly on (generalized) linear models. Bayesian additive regression trees (BART), in contrast, is a reliable method to obtain high-quality nonlinear out of sample predictions without overfitting. Unfortunately, the default prior from BART may be too inflexible to handle sparse situations where the number of predictors approaches or surpasses the number of observations. Motivated by our real data application, this article proposes an alternative prior based on the logit normal distribution because it provides a framework that is adaptive to sparsity and can model informative functional annotations. It also provides a framework to incorporate prior information about the between SNP correlations. Computational details for carrying out inference are presented along with the results from a simulation study and a genome-wide prediction analysis of the Alzheimer's Disease Neuroimaging Initiative data.</p>","PeriodicalId":12710,"journal":{"name":"Genetic Epidemiology","volume":"47 1","pages":"26-44"},"PeriodicalIF":1.7000,"publicationDate":"2022-11-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/gepi.22505","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetic Epidemiology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/gepi.22505","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
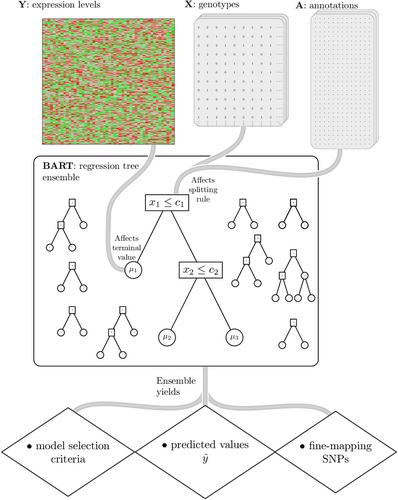
Using high-dimensional genetic variants such as single nucleotide polymorphisms (SNP) to predict complex diseases and traits has important applications in basic research and other clinical settings. For example, predicting gene expression is a necessary first step to identify (putative) causal genes in transcriptome-wide association studies. Due to weak signals, high-dimensionality, and linkage disequilibrium (correlation) among SNPs, building such a prediction model is challenging. However, functional annotations at the SNP level (e.g., as epigenomic data across multiple cell- or tissue-types) are available and could be used to inform predictor importance and aid in outcome prediction. Existing approaches to incorporate annotations have been based mainly on (generalized) linear models. Bayesian additive regression trees (BART), in contrast, is a reliable method to obtain high-quality nonlinear out of sample predictions without overfitting. Unfortunately, the default prior from BART may be too inflexible to handle sparse situations where the number of predictors approaches or surpasses the number of observations. Motivated by our real data application, this article proposes an alternative prior based on the logit normal distribution because it provides a framework that is adaptive to sparsity and can model informative functional annotations. It also provides a framework to incorporate prior information about the between SNP correlations. Computational details for carrying out inference are presented along with the results from a simulation study and a genome-wide prediction analysis of the Alzheimer's Disease Neuroimaging Initiative data.
期刊介绍:
Genetic Epidemiology is a peer-reviewed journal for discussion of research on the genetic causes of the distribution of human traits in families and populations. Emphasis is placed on the relative contribution of genetic and environmental factors to human disease as revealed by genetic, epidemiological, and biologic investigations.
Genetic Epidemiology primarily publishes papers in statistical genetics, a research field that is primarily concerned with development of statistical, bioinformatical, and computational models for analyzing genetic data. Incorporation of underlying biology and population genetics into conceptual models is favored. The Journal seeks original articles comprising either applied research or innovative statistical, mathematical, computational, or genomic methodologies that advance studies in genetic epidemiology. Other types of reports are encouraged, such as letters to the editor, topic reviews, and perspectives from other fields of research that will likely enrich the field of genetic epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


