Philippe Pinel, Gwenn Guichaoua, Matthieu Najm, Stéphanie Labouille, Nicolas Drizard, Yann Gaston-Mathé, Brice Hoffmann, Véronique Stoven
{"title":"Exploring isofunctional molecules: Design of a benchmark and evaluation of prediction performance.","authors":"Philippe Pinel, Gwenn Guichaoua, Matthieu Najm, Stéphanie Labouille, Nicolas Drizard, Yann Gaston-Mathé, Brice Hoffmann, Véronique Stoven","doi":"10.1002/minf.202200216","DOIUrl":null,"url":null,"abstract":"<p><p>Identification of novel chemotypes with biological activity similar to a known active molecule is an important challenge in drug discovery called 'scaffold hopping'. Small-, medium-, and large-step scaffold hopping efforts may lead to increasing degrees of chemical structure novelty with respect to the parent compound. In the present paper, we focus on the problem of large-step scaffold hopping. We assembled a high quality and well characterized dataset of scaffold hopping examples comprising pairs of active molecules and including a variety of protein targets. This dataset was used to build a benchmark corresponding to the setting of real-life applications: one active molecule is known, and the second active is searched among a set of decoys chosen in a way to avoid statistical bias. This allowed us to evaluate the performance of computational methods for solving large-step scaffold hopping problems. In particular, we assessed how difficult these problems are, particularly for classical 2D and 3D ligand-based methods. We also showed that a machine-learning chemogenomic algorithm outperforms classical methods and we provided some useful hints for future improvements.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":null,"pages":null},"PeriodicalIF":2.8000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200216","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
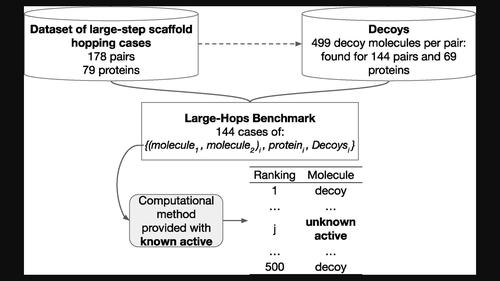
Identification of novel chemotypes with biological activity similar to a known active molecule is an important challenge in drug discovery called 'scaffold hopping'. Small-, medium-, and large-step scaffold hopping efforts may lead to increasing degrees of chemical structure novelty with respect to the parent compound. In the present paper, we focus on the problem of large-step scaffold hopping. We assembled a high quality and well characterized dataset of scaffold hopping examples comprising pairs of active molecules and including a variety of protein targets. This dataset was used to build a benchmark corresponding to the setting of real-life applications: one active molecule is known, and the second active is searched among a set of decoys chosen in a way to avoid statistical bias. This allowed us to evaluate the performance of computational methods for solving large-step scaffold hopping problems. In particular, we assessed how difficult these problems are, particularly for classical 2D and 3D ligand-based methods. We also showed that a machine-learning chemogenomic algorithm outperforms classical methods and we provided some useful hints for future improvements.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


