David A. A. Baranger, Alexander S. Hatoum, Renato Polimanti, Joel Gelernter, Howard J. Edenberg, Ryan Bogdan, Arpana Agrawal
{"title":"Multi-omics cannot replace sample size in genome-wide association studies","authors":"David A. A. Baranger, Alexander S. Hatoum, Renato Polimanti, Joel Gelernter, Howard J. Edenberg, Ryan Bogdan, Arpana Agrawal","doi":"10.1111/gbb.12846","DOIUrl":null,"url":null,"abstract":"<p>The integration of multi-omics information (e.g., epigenetics and transcriptomics) can be useful for interpreting findings from genome-wide association studies (GWAS). It has been suggested that multi-omics could circumvent or greatly reduce the need to increase GWAS sample sizes for novel variant discovery. We tested whether incorporating multi-omics information in earlier and smaller-sized GWAS boosts true-positive discovery of genes that were later revealed by larger GWAS of the same/similar traits. We applied 10 different analytic approaches to integrating multi-omics data from 12 sources (e.g., Genotype-Tissue Expression project) to test whether earlier and smaller GWAS of 4 brain-related traits (alcohol use disorder/problematic alcohol use, major depression/depression, schizophrenia, and intracranial volume/brain volume) could detect genes that were revealed by a later and larger GWAS. Multi-omics data did not reliably identify novel genes in earlier less-powered GWAS (PPV <0.2; 80% false-positive associations). Machine learning predictions marginally increased the number of identified novel genes, correctly identifying 1–8 additional genes, but only for well-powered early GWAS of highly heritable traits (i.e., intracranial volume and schizophrenia). Although multi-omics, particularly positional mapping (i.e., fastBAT, MAGMA, and H-MAGMA), can help to prioritize genes within genome-wide significant loci (PPVs = 0.5–1.0) and translate them into information about disease biology, it does not reliably increase novel gene discovery in brain-related GWAS. To increase power for discovery of novel genes and loci, increasing sample size is required.</p>","PeriodicalId":50426,"journal":{"name":"Genes Brain and Behavior","volume":"22 6","pages":""},"PeriodicalIF":2.3000,"publicationDate":"2023-03-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/gbb.12846","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genes Brain and Behavior","FirstCategoryId":"102","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/gbb.12846","RegionNum":4,"RegionCategory":"心理学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BEHAVIORAL SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
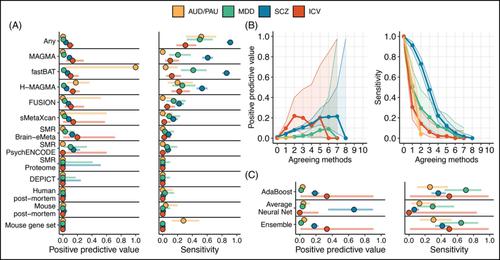
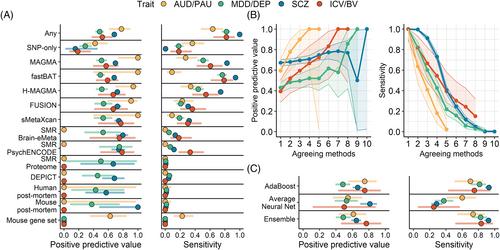
The integration of multi-omics information (e.g., epigenetics and transcriptomics) can be useful for interpreting findings from genome-wide association studies (GWAS). It has been suggested that multi-omics could circumvent or greatly reduce the need to increase GWAS sample sizes for novel variant discovery. We tested whether incorporating multi-omics information in earlier and smaller-sized GWAS boosts true-positive discovery of genes that were later revealed by larger GWAS of the same/similar traits. We applied 10 different analytic approaches to integrating multi-omics data from 12 sources (e.g., Genotype-Tissue Expression project) to test whether earlier and smaller GWAS of 4 brain-related traits (alcohol use disorder/problematic alcohol use, major depression/depression, schizophrenia, and intracranial volume/brain volume) could detect genes that were revealed by a later and larger GWAS. Multi-omics data did not reliably identify novel genes in earlier less-powered GWAS (PPV <0.2; 80% false-positive associations). Machine learning predictions marginally increased the number of identified novel genes, correctly identifying 1–8 additional genes, but only for well-powered early GWAS of highly heritable traits (i.e., intracranial volume and schizophrenia). Although multi-omics, particularly positional mapping (i.e., fastBAT, MAGMA, and H-MAGMA), can help to prioritize genes within genome-wide significant loci (PPVs = 0.5–1.0) and translate them into information about disease biology, it does not reliably increase novel gene discovery in brain-related GWAS. To increase power for discovery of novel genes and loci, increasing sample size is required.
期刊介绍:
Genes, Brain and Behavior was launched in 2002 with the aim of publishing top quality research in behavioral and neural genetics in their broadest sense. The emphasis is on the analysis of the behavioral and neural phenotypes under consideration, the unifying theme being the genetic approach as a tool to increase our understanding of these phenotypes.
Genes Brain and Behavior is pleased to offer the following features:
8 issues per year
online submissions with first editorial decisions within 3-4 weeks and fast publication at Wiley-Blackwells
High visibility through its coverage by PubMed/Medline, Current Contents and other major abstracting and indexing services
Inclusion in the Wiley-Blackwell consortial license, extending readership to thousands of international libraries and institutions
A large and varied editorial board comprising of international specialists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


