MOLASS: Software for automatic processing of matrix data obtained from small-angle X-ray scattering and UV-visible spectroscopy combined with size-exclusion chromatography.
{"title":"MOLASS: Software for automatic processing of matrix data obtained from small-angle X-ray scattering and UV-visible spectroscopy combined with size-exclusion chromatography.","authors":"Kento Yonezawa, Masatsuyo Takahashi, Keiko Yatabe, Yasuko Nagatani, Nobutaka Shimizu","doi":"10.2142/biophysico.bppb-v20.0001","DOIUrl":null,"url":null,"abstract":"<p><p>Recent small-angle X-ray scattering (SAXS) for biological macromolecules (BioSAXS) is generally combined with size-exclusion chromatography (SEC-SAXS) at synchrotron facilities worldwide. For SEC-SAXS analysis, the final scattering profile for the target molecule is calculated from a large volume of continuously collected data. It would be ideal to automate this process; however, several complex problems exist regarding data measurement and analysis that have prevented automation. Here, we developed the analytical software MOLASS (Matrix Optimization with Low-rank factorization for Automated analysis of SEC-SAXS) to automatically calculate the final scattering profiles for solution structure analysis of target molecules. In this paper, the strategies for automatic analysis of SEC-SAXS data are described, including correction of baseline-drift using a low percentile method, optimization of peak decompositions composed of multiple scattering components using modified Gaussian fitting against the chromatogram, and rank determination for extrapolation to infinite dilution. In order to easily calculate each scattering component, the Moore-Penrose pseudo-inverse matrix is adopted as a basic calculation. Furthermore, this analysis method, in combination with UV-visible spectroscopy, led to better results in terms of accuracy in peak decomposition. Therefore, MOLASS will be able to smoothly suggest to users an accurate scattering profile for the subsequent structural analysis.</p>","PeriodicalId":8976,"journal":{"name":"Biophysics and Physicobiology","volume":"20 1","pages":"e200001"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6c/80/20_e200001.PMC10203098.pdf","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysics and Physicobiology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2142/biophysico.bppb-v20.0001","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
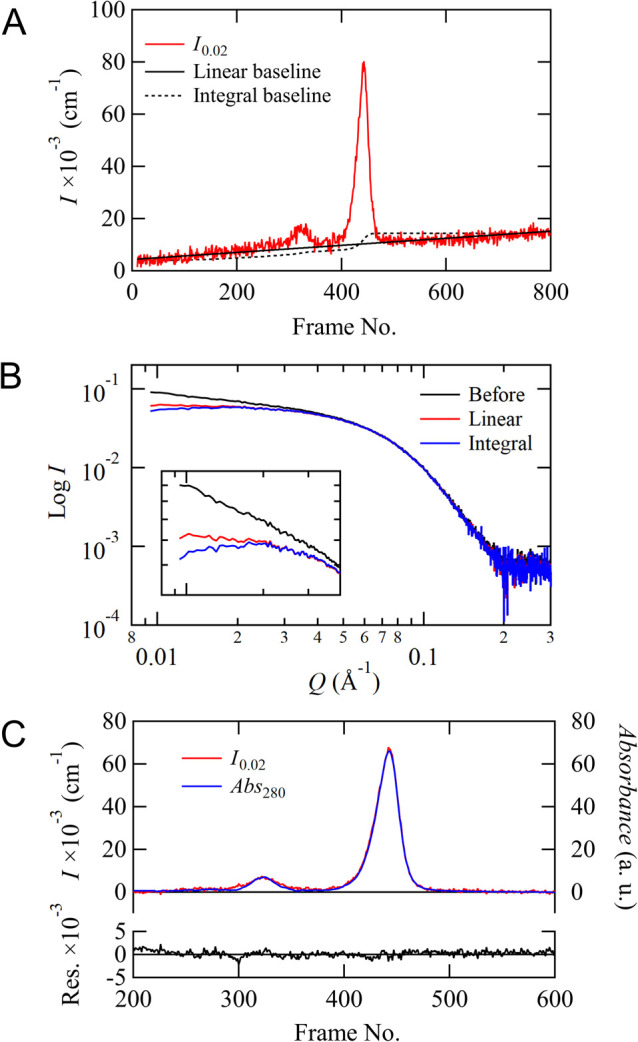
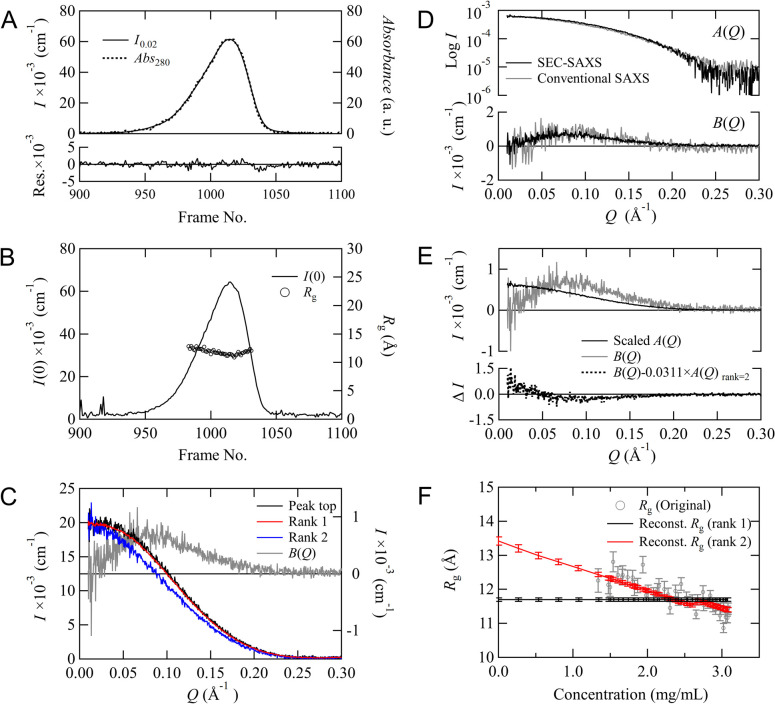
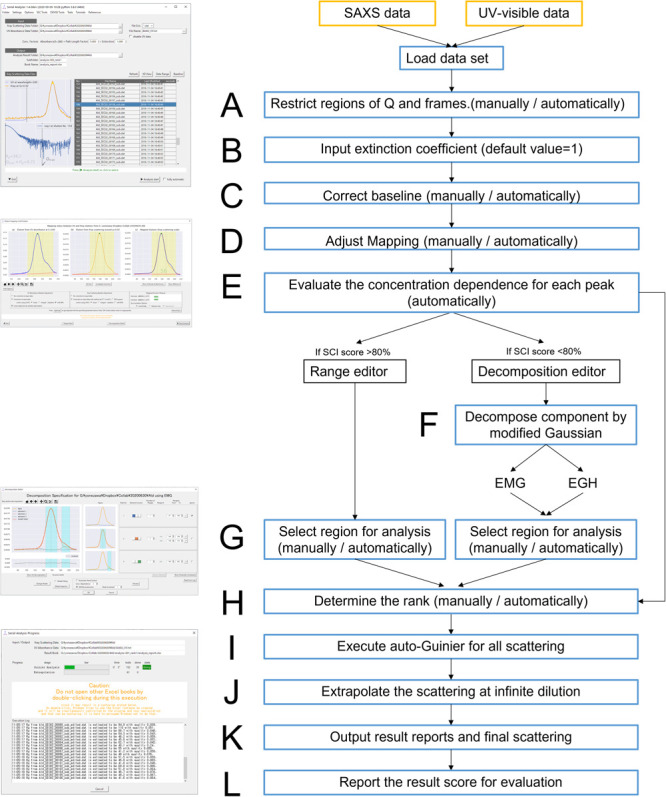
Recent small-angle X-ray scattering (SAXS) for biological macromolecules (BioSAXS) is generally combined with size-exclusion chromatography (SEC-SAXS) at synchrotron facilities worldwide. For SEC-SAXS analysis, the final scattering profile for the target molecule is calculated from a large volume of continuously collected data. It would be ideal to automate this process; however, several complex problems exist regarding data measurement and analysis that have prevented automation. Here, we developed the analytical software MOLASS (Matrix Optimization with Low-rank factorization for Automated analysis of SEC-SAXS) to automatically calculate the final scattering profiles for solution structure analysis of target molecules. In this paper, the strategies for automatic analysis of SEC-SAXS data are described, including correction of baseline-drift using a low percentile method, optimization of peak decompositions composed of multiple scattering components using modified Gaussian fitting against the chromatogram, and rank determination for extrapolation to infinite dilution. In order to easily calculate each scattering component, the Moore-Penrose pseudo-inverse matrix is adopted as a basic calculation. Furthermore, this analysis method, in combination with UV-visible spectroscopy, led to better results in terms of accuracy in peak decomposition. Therefore, MOLASS will be able to smoothly suggest to users an accurate scattering profile for the subsequent structural analysis.
目前,生物大分子的小角度x射线散射(SAXS)技术在世界范围内的同步加速器上普遍采用与粒径隔离色谱(SEC-SAXS)相结合的方法。对于SEC-SAXS分析,从大量连续收集的数据中计算目标分子的最终散射曲线。将这一过程自动化是理想的;然而,在数据测量和分析方面存在一些复杂的问题,这些问题阻碍了自动化。在此,我们开发了分析软件MOLASS (Matrix Optimization with Low-rank factorization for Automated analysis of SEC-SAXS),用于自动计算目标分子溶液结构分析的最终散射曲线。本文介绍了SEC-SAXS数据自动分析的策略,包括使用低百分位法校正基线漂移,使用改进的高斯拟合针对色谱图优化由多个散射分量组成的峰分解,以及用于无限稀释外推的等级确定。为了方便计算各个散射分量,采用Moore-Penrose伪逆矩阵作为基本计算。此外,该分析方法与紫外-可见光谱相结合,在峰分解的准确性方面取得了更好的结果。因此,MOLASS将能够顺利地向用户提供准确的散射剖面,用于后续的结构分析。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



