Milder presentation of osteogenesis imperfecta type VIII due to compound heterozygosity for a predicted loss-of-function variant and novel missense variant in P3H1-further expansion of the phenotypic spectrum.
Kristen A Mikhail, Elizabeth VanSickle, Linda Z Rossetti
{"title":"Milder presentation of osteogenesis imperfecta type VIII due to compound heterozygosity for a predicted loss-of-function variant and novel missense variant in <i>P3H1</i>-further expansion of the phenotypic spectrum.","authors":"Kristen A Mikhail, Elizabeth VanSickle, Linda Z Rossetti","doi":"10.1101/mcs.a006260","DOIUrl":null,"url":null,"abstract":"<p><p>Osteogenesis imperfecta (OI) is a heritable disorder of bone metabolism characterized by multiple fractures with minimal trauma. Autosomal recessive OI type VIII is associated with biallelic pathogenic variants in <i>P3H1</i> and classically characterized by skeletal anomalies in addition to significant bone fragility, sometimes presenting with in utero fractures and/or neonatal lethality. <i>P3H1</i> encodes a collagen prolyl hydroxylase that critically 3-hydroxylates proline residue 986 on the α chain of collagen types I and II to achieve proper folding and assembly of mature collagen and is present in a complex with CRTAP and CypB. Most individuals with OI type VIII have had biallelic predicted loss-of-function variants leading to reduced or absent levels of <i>P3H1</i> mRNA. The reported missense variants have all fallen in the catalytic domain of the protein and are thought to be associated with a milder phenotype. Here, we describe an infant presenting with five long bone fractures in the first year of life found to have a novel missense variant in <i>trans</i> with a nonsense variant in <i>P3H1</i> without any other bony anomalies on imaging. We hypothesize that missense variants in the catalytic domain of P3H1 lead to decreased but not absent hydroxylation of Pro986, with preserved KDEL retention signal and complex stability, causing an attenuated phenotype.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":"9 1","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/2a/MCS006260Mik.PMC10111797.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006260","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
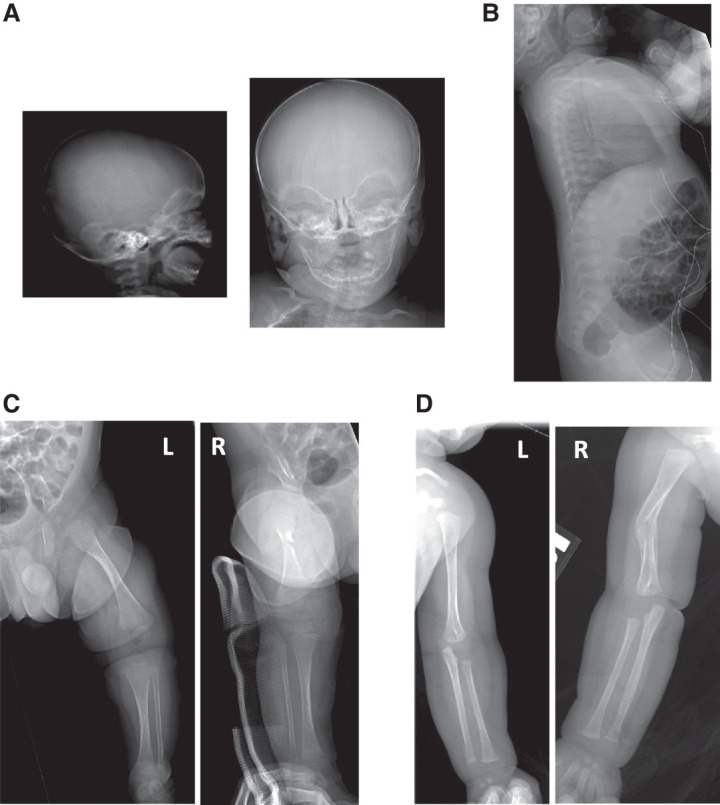
Osteogenesis imperfecta (OI) is a heritable disorder of bone metabolism characterized by multiple fractures with minimal trauma. Autosomal recessive OI type VIII is associated with biallelic pathogenic variants in P3H1 and classically characterized by skeletal anomalies in addition to significant bone fragility, sometimes presenting with in utero fractures and/or neonatal lethality. P3H1 encodes a collagen prolyl hydroxylase that critically 3-hydroxylates proline residue 986 on the α chain of collagen types I and II to achieve proper folding and assembly of mature collagen and is present in a complex with CRTAP and CypB. Most individuals with OI type VIII have had biallelic predicted loss-of-function variants leading to reduced or absent levels of P3H1 mRNA. The reported missense variants have all fallen in the catalytic domain of the protein and are thought to be associated with a milder phenotype. Here, we describe an infant presenting with five long bone fractures in the first year of life found to have a novel missense variant in trans with a nonsense variant in P3H1 without any other bony anomalies on imaging. We hypothesize that missense variants in the catalytic domain of P3H1 lead to decreased but not absent hydroxylation of Pro986, with preserved KDEL retention signal and complex stability, causing an attenuated phenotype.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


