Joint Secondary Transcriptomic Analysis of Non-Hodgkin's B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers.
Naomi Rapier-Sharman, Jeffrey Clancy, Brett E Pickett
{"title":"Joint Secondary Transcriptomic Analysis of Non-Hodgkin's B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers.","authors":"Naomi Rapier-Sharman, Jeffrey Clancy, Brett E Pickett","doi":"10.26502/jbsb.5107040","DOIUrl":null,"url":null,"abstract":"<p><p>Approximately 450,000 cases of Non-Hodgkin's lymphoma are annually diagnosed worldwide, resulting in ~240,000 deaths. An augmented understanding of the common mechanisms of pathology among larger numbers of B-cell Non-Hodgkin's Lymphoma (BCNHL) patients is sorely needed. We consequently performed a large joint secondary transcriptomic analysis of the available BCNHL RNA-sequencing projects from GEO, consisting of 322 relevant samples across ten distinct public studies, to find common underlying mechanisms and biomarkers across multiple BCNHL subtypes and patient subpopulations; limitations may include lack of diversity in certain ethnicities and age groups and limited clinical subtype diversity due to sample availability. We found ~10,400 significant differentially expressed genes (FDR-adjusted p-value < 0.05) and 33 significantly modulated pathways (Bonferroni-adjusted p-value < 0.05) when comparing BCNHL samples to non-diseased B-cell samples. Our findings included a significant class of proteoglycans not previously associated with lymphomas as well as significant modulation of genes that code for extracellular matrix-associated proteins. Our drug repurposing analysis predicted new candidates for repurposed drugs including ocriplasmin and collagenase. We also used a machine learning approach to identify robust BCNHL biomarkers that include YES1, FERMT2, and FAM98B, which have not previously been associated with BCNHL in the literature, but together provide ~99.9% combined specificity and sensitivity for differentiating lymphoma cells from healthy B-cells based on measurement of transcript expression levels in B-cells. This analysis supports past findings and validates existing knowledge while providing novel insights into the inner workings and mechanisms of transformed B-cell lymphomas that could give rise to improved diagnostics and/or therapeutics.</p>","PeriodicalId":73617,"journal":{"name":"Journal of bioinformatics and systems biology : Open access","volume":"5 4","pages":"119-135"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9980876/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of bioinformatics and systems biology : Open access","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.26502/jbsb.5107040","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/27 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
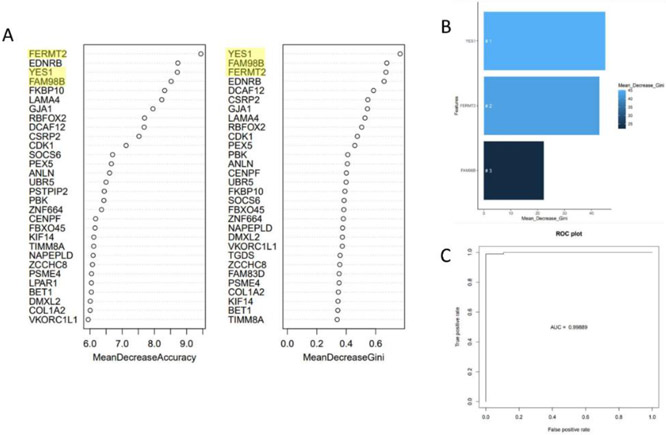
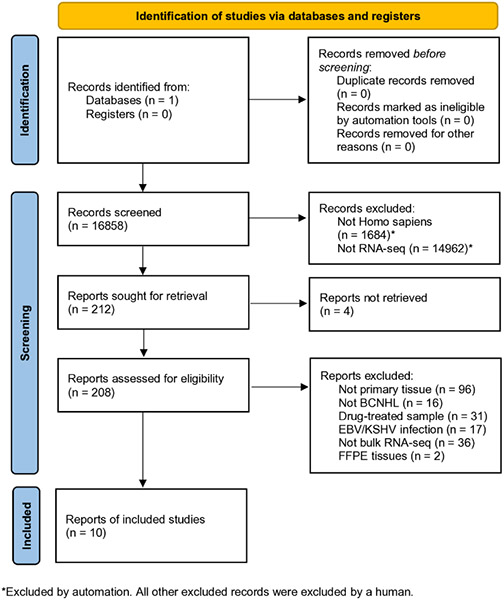
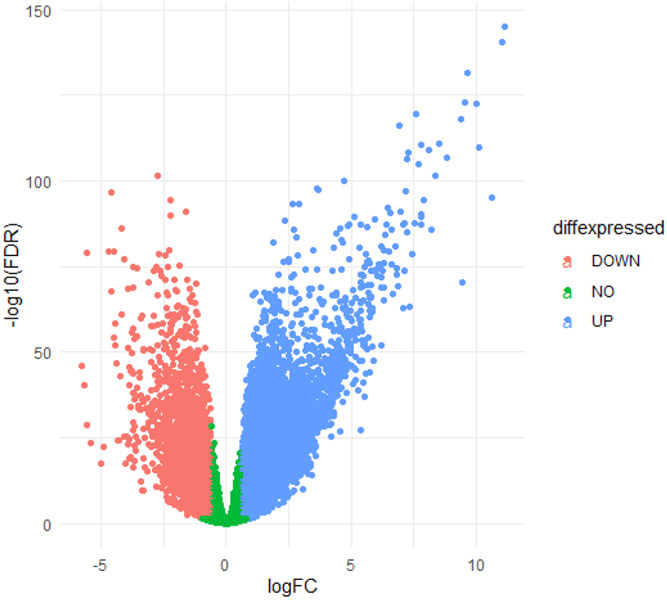
Approximately 450,000 cases of Non-Hodgkin's lymphoma are annually diagnosed worldwide, resulting in ~240,000 deaths. An augmented understanding of the common mechanisms of pathology among larger numbers of B-cell Non-Hodgkin's Lymphoma (BCNHL) patients is sorely needed. We consequently performed a large joint secondary transcriptomic analysis of the available BCNHL RNA-sequencing projects from GEO, consisting of 322 relevant samples across ten distinct public studies, to find common underlying mechanisms and biomarkers across multiple BCNHL subtypes and patient subpopulations; limitations may include lack of diversity in certain ethnicities and age groups and limited clinical subtype diversity due to sample availability. We found ~10,400 significant differentially expressed genes (FDR-adjusted p-value < 0.05) and 33 significantly modulated pathways (Bonferroni-adjusted p-value < 0.05) when comparing BCNHL samples to non-diseased B-cell samples. Our findings included a significant class of proteoglycans not previously associated with lymphomas as well as significant modulation of genes that code for extracellular matrix-associated proteins. Our drug repurposing analysis predicted new candidates for repurposed drugs including ocriplasmin and collagenase. We also used a machine learning approach to identify robust BCNHL biomarkers that include YES1, FERMT2, and FAM98B, which have not previously been associated with BCNHL in the literature, but together provide ~99.9% combined specificity and sensitivity for differentiating lymphoma cells from healthy B-cells based on measurement of transcript expression levels in B-cells. This analysis supports past findings and validates existing knowledge while providing novel insights into the inner workings and mechanisms of transformed B-cell lymphomas that could give rise to improved diagnostics and/or therapeutics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


