A Sarajlija, L Armengol, A Maver, I Kitic, D Prokic, M Cehic, M S Djuricic, B Peterlin
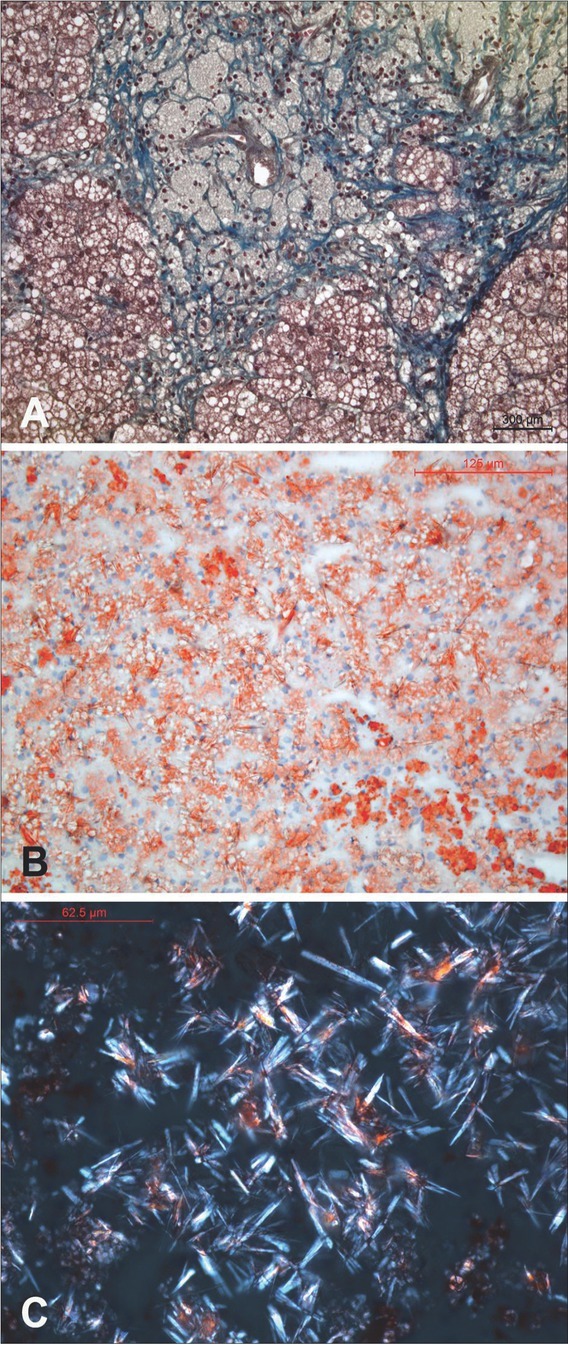
{"title":"A Novel Variant in the <i>LIPA</i> Gene Associated with Distinct Phenotype.","authors":"A Sarajlija, L Armengol, A Maver, I Kitic, D Prokic, M Cehic, M S Djuricic, B Peterlin","doi":"10.2478/bjmg-2022-0010","DOIUrl":null,"url":null,"abstract":"<p><p>Deficiency of lysosomal acid lipase (LAL-D) is caused by biallelic pathogenic variants in the <i>LIPA</i> gene. Spectrum of LAL-D ranges from early onset of hepatosplenomegaly and psychomotor regression (Wolman disease) to a more chronic course (cholesteryl ester storage disease - CESD). The diagnosis is based on lipid and biomarker profiles, specific liver histopathology, enzyme deficiency, and identification of causative genetic variants. Biomarker findings are a useful for diagnostics of LAL-D, including high plasma concentration of chitotriosidase as well as elevated oxysterols. Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation, and stem cell transplantation. We present two pairs of siblings from Serbia with a distinctive phenotype resembling LAL-D with a novel variant of unknown significance (VUS) detected in the <i>LIPA</i> gene and residual LAL activity. All patients presented with hepatosplenomegaly at early childhood. In siblings from family 1, compound heterozygosity for a pathogenic c.419G>A (p.Trp140Ter) variant and a novel VUS c.851C>T (p.Ser284Phe) was detected. Patients from family 2 were homozygous for c.851C>T VUS and both have typical histopathologic findings for LAL-D in the liver. Enzyme activity of LAL was tested in three patients and reported as sufficient, and therefore enzyme replacement therapy could not be approved. When confronted with a challenge of diagnosing an inherited metabolic disorder, several aspects are taken into consideration: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. This report brings cases to light which have a considerable discrepancy between those aspects, namely the preserved LAL enzyme activity in presence of clinical manifestations and rare variants in the <i>LIPA</i> gene.</p>","PeriodicalId":55403,"journal":{"name":"Balkan Journal of Medical Genetics","volume":"25 1","pages":"93-100"},"PeriodicalIF":0.9000,"publicationDate":"2022-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0d/f0/bjmg-25-093.PMC9985358.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2022-0010","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Deficiency of lysosomal acid lipase (LAL-D) is caused by biallelic pathogenic variants in the LIPA gene. Spectrum of LAL-D ranges from early onset of hepatosplenomegaly and psychomotor regression (Wolman disease) to a more chronic course (cholesteryl ester storage disease - CESD). The diagnosis is based on lipid and biomarker profiles, specific liver histopathology, enzyme deficiency, and identification of causative genetic variants. Biomarker findings are a useful for diagnostics of LAL-D, including high plasma concentration of chitotriosidase as well as elevated oxysterols. Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation, and stem cell transplantation. We present two pairs of siblings from Serbia with a distinctive phenotype resembling LAL-D with a novel variant of unknown significance (VUS) detected in the LIPA gene and residual LAL activity. All patients presented with hepatosplenomegaly at early childhood. In siblings from family 1, compound heterozygosity for a pathogenic c.419G>A (p.Trp140Ter) variant and a novel VUS c.851C>T (p.Ser284Phe) was detected. Patients from family 2 were homozygous for c.851C>T VUS and both have typical histopathologic findings for LAL-D in the liver. Enzyme activity of LAL was tested in three patients and reported as sufficient, and therefore enzyme replacement therapy could not be approved. When confronted with a challenge of diagnosing an inherited metabolic disorder, several aspects are taken into consideration: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. This report brings cases to light which have a considerable discrepancy between those aspects, namely the preserved LAL enzyme activity in presence of clinical manifestations and rare variants in the LIPA gene.
期刊介绍:
Balkan Journal of Medical Genetics is a journal in the English language for publication of articles involving all branches of medical genetics: human cytogenetics, molecular genetics, clinical genetics, immunogenetics, oncogenetics, pharmacogenetics, population genetics, genetic screening and diagnosis of monogenic and polygenic diseases, prenatal and preimplantation genetic diagnosis, genetic counselling, advances in treatment and prevention.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


