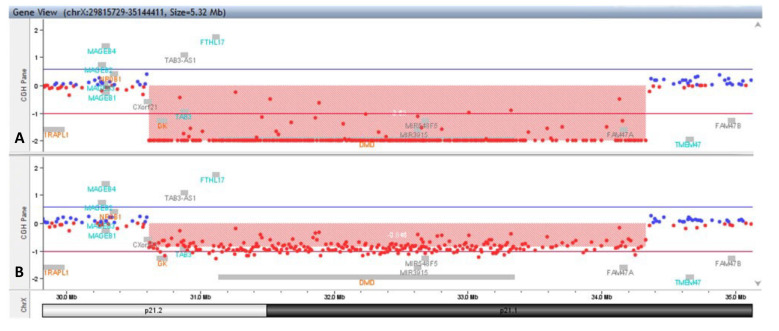
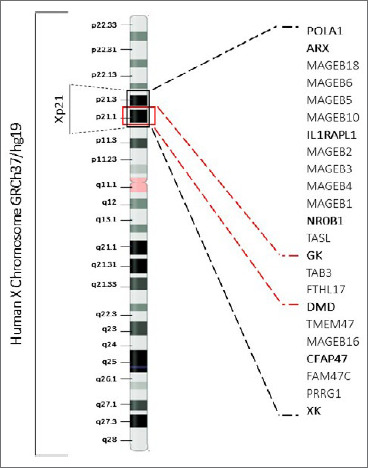
Xp21 contiguous gene deletion syndrome presenting as Duchenne muscular dystrophy and glycerol kinase deficiency associated with intellectual disability: case report and review literature.
Antonella Pizza, Esther Picillo, Maria Elena Onore, Marianna Scutifero, Luigia Passamano, Vincenzo Nigro, Luisa Politano
{"title":"Xp21 contiguous gene deletion syndrome presenting as Duchenne muscular dystrophy and glycerol kinase deficiency associated with intellectual disability: case report and review literature.","authors":"Antonella Pizza, Esther Picillo, Maria Elena Onore, Marianna Scutifero, Luigia Passamano, Vincenzo Nigro, Luisa Politano","doi":"10.36185/2532-1900-246","DOIUrl":null,"url":null,"abstract":"<p><p>The contiguous gene deletion syndromes (CGDS) are rare genomic disorders resulting from the deletion of large segments of DNA, manifested as the concurrence of apparently unrelated clinical features. A typical example of CGDS is Xp21 contiguous gene deletion syndrome that involves <i>GK</i> and its neigh-boring genes (usually <i>DMD</i> and <i>NR0B1</i>) and results in a complex phenotype, which is related to the size of deletion and involved genes. Development delay and intellectual disability are almost a constant feature of patients with CGDS. We report the case of a boy with Duchenne muscular dystrophy (DMD) and glycerol kinase deficiency (GKD) as part of the contiguous gene deletion syndrome Xp2.1, in association with intellectual disability (ID) in whom multiplex ligation-dependent probe amplification (MLPA) test first identified a hemizygous deletion involving the entire dystrophin gene. Subsequently, the array CGH study identified a maternally inherited hemizygous deletion of the Xp21.2-Xp21.1 region of approximately 3.7Mb that included both <i>DMD</i> and <i>GK</i> genes confirming the diagnosis of Xp21 CGDS. Moreover, we report a review of the cases published in the literature over the last 20 years, for which a better description of the genes involved in the syndrome was available. Intellectual disability does not appear as a constant feature of the syndrome, reiterating the concept that complex GKD syndrome results from small deletions that affect closely related but separate loci for DMD, GK and adrenal hypoplasia, rather than a single large deletion including all genes. This case highlights the importance of more in-depth genetic investigations in presence of apparently unrelated clinical findings, allowing an accurate diagnosis of contiguous gene deletion syndromes.</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"42 1","pages":"24-30"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/a4/00/am-2023-01-24.PMC10115399.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-246","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
The contiguous gene deletion syndromes (CGDS) are rare genomic disorders resulting from the deletion of large segments of DNA, manifested as the concurrence of apparently unrelated clinical features. A typical example of CGDS is Xp21 contiguous gene deletion syndrome that involves GK and its neigh-boring genes (usually DMD and NR0B1) and results in a complex phenotype, which is related to the size of deletion and involved genes. Development delay and intellectual disability are almost a constant feature of patients with CGDS. We report the case of a boy with Duchenne muscular dystrophy (DMD) and glycerol kinase deficiency (GKD) as part of the contiguous gene deletion syndrome Xp2.1, in association with intellectual disability (ID) in whom multiplex ligation-dependent probe amplification (MLPA) test first identified a hemizygous deletion involving the entire dystrophin gene. Subsequently, the array CGH study identified a maternally inherited hemizygous deletion of the Xp21.2-Xp21.1 region of approximately 3.7Mb that included both DMD and GK genes confirming the diagnosis of Xp21 CGDS. Moreover, we report a review of the cases published in the literature over the last 20 years, for which a better description of the genes involved in the syndrome was available. Intellectual disability does not appear as a constant feature of the syndrome, reiterating the concept that complex GKD syndrome results from small deletions that affect closely related but separate loci for DMD, GK and adrenal hypoplasia, rather than a single large deletion including all genes. This case highlights the importance of more in-depth genetic investigations in presence of apparently unrelated clinical findings, allowing an accurate diagnosis of contiguous gene deletion syndromes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


