Xuyi Zhan, Shaoyin Bao, Xumei Li, Shaojun Zhou, Maha Raja Dahar, Nengming Lin, Xiugui Chen, Chengshan Niu, Kaige Ji, Yusheng Wu, Kui Zeng, Zhihua Tang, Lushan Yu
{"title":"Metabolism and pharmacokinetic study of deuterated osimertinib","authors":"Xuyi Zhan, Shaoyin Bao, Xumei Li, Shaojun Zhou, Maha Raja Dahar, Nengming Lin, Xiugui Chen, Chengshan Niu, Kaige Ji, Yusheng Wu, Kui Zeng, Zhihua Tang, Lushan Yu","doi":"10.1002/bdd.2347","DOIUrl":null,"url":null,"abstract":"<p>Osimertinib is a highly selective third-generation irreversible inhibitor of epidermal growth factor receptor mutant, which can be utilized to treat non-small cell lung cancer. As the substrate of cytochrome P450 enzyme, it is mainly metabolized by the CYP3A enzyme in humans. Among the metabolites produced by osimertinib, AZ5104, and AZ7550, which are demethylated that is most vital. Nowadays, deuteration is a new design approach for several drugs. This popular strategy is deemed to improve the pharmacokinetic characteristics of the original drugs. Therefore, in this study the metabolism profiles of osimertinib and its deuterated compound (osimertinib-d3) in liver microsomes and human recombinant cytochrome P450 isoenzymes and the pharmacokinetics in rats and humans were compared. After deuteration, its kinetic isotope effect greatly inhibited the metabolic pathway that produces AZ5104. The plasma concentration of the key metabolite AZ5104 of osimertinib-d3 in rats and humans decreased significantly compared with that of the osimertinib. This phenomenon was consistent with the results of the metabolism studies in vitro. In addition, the in vivo results indicated that osimertinib-d3 had higher systemic exposure (AUC) and peak concentration (C<sub>max</sub>) compared with the osimertinib in rats and human body.</p>","PeriodicalId":8865,"journal":{"name":"Biopharmaceutics & Drug Disposition","volume":"44 2","pages":"165-174"},"PeriodicalIF":2.0000,"publicationDate":"2023-01-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biopharmaceutics & Drug Disposition","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bdd.2347","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
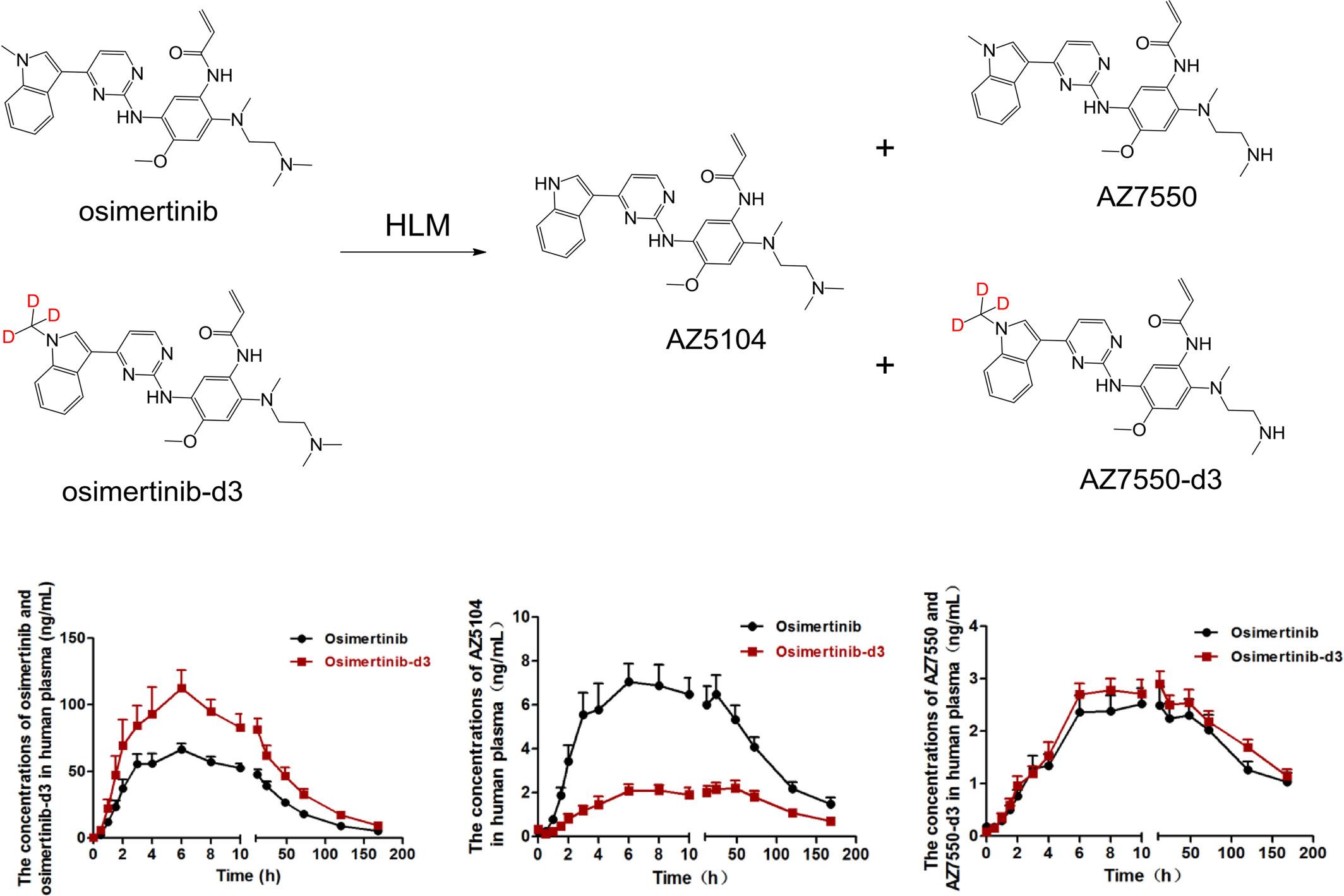
Osimertinib is a highly selective third-generation irreversible inhibitor of epidermal growth factor receptor mutant, which can be utilized to treat non-small cell lung cancer. As the substrate of cytochrome P450 enzyme, it is mainly metabolized by the CYP3A enzyme in humans. Among the metabolites produced by osimertinib, AZ5104, and AZ7550, which are demethylated that is most vital. Nowadays, deuteration is a new design approach for several drugs. This popular strategy is deemed to improve the pharmacokinetic characteristics of the original drugs. Therefore, in this study the metabolism profiles of osimertinib and its deuterated compound (osimertinib-d3) in liver microsomes and human recombinant cytochrome P450 isoenzymes and the pharmacokinetics in rats and humans were compared. After deuteration, its kinetic isotope effect greatly inhibited the metabolic pathway that produces AZ5104. The plasma concentration of the key metabolite AZ5104 of osimertinib-d3 in rats and humans decreased significantly compared with that of the osimertinib. This phenomenon was consistent with the results of the metabolism studies in vitro. In addition, the in vivo results indicated that osimertinib-d3 had higher systemic exposure (AUC) and peak concentration (Cmax) compared with the osimertinib in rats and human body.
期刊介绍:
Biopharmaceutics & Drug Dispositionpublishes original review articles, short communications, and reports in biopharmaceutics, drug disposition, pharmacokinetics and pharmacodynamics, especially those that have a direct relation to the drug discovery/development and the therapeutic use of drugs. These includes:
- animal and human pharmacological studies that focus on therapeutic response. pharmacodynamics, and toxicity related to plasma and tissue concentrations of drugs and their metabolites,
- in vitro and in vivo drug absorption, distribution, metabolism, transport, and excretion studies that facilitate investigations related to the use of drugs in man
- studies on membrane transport and enzymes, including their regulation and the impact of pharmacogenomics on drug absorption and disposition,
- simulation and modeling in drug discovery and development
- theoretical treatises
- includes themed issues and reviews
and exclude manuscripts on
- bioavailability studies reporting only on simple PK parameters such as Cmax, tmax and t1/2 without mechanistic interpretation
- analytical methods

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


