{"title":"Nicorandil protects podocytes via modulation of antioxidative capacity in acute puromycin aminonucleoside-induced nephrosis in rats.","authors":"Masaki Yamanaka, Yoshifuru Tamura, Emiko Kuribayashi-Okuma, Shunya Uchida, Shigeru Shibata","doi":"10.1152/ajprenal.00144.2022","DOIUrl":null,"url":null,"abstract":"<p><p>Nephrotic syndrome, characterized by proteinuria and hypoalbuminemia, results from the dysregulation of glomerular podocytes and is a significant cause of end-stage kidney disease. Patients with idiopathic nephrotic syndrome are generally treated with immunosuppressive agents; however, these agents produce various adverse effects. Previously, we reported the renoprotective effects of a stimulator of the mitochondrial ATP-dependent K<sup>+</sup> channel (MitK<sub>ATP</sub>), nicorandil, in a remnant kidney model. Nonetheless, the cellular targets of these effects remain unknown. Here, we examined the effect of nicorandil on puromycin aminonucleoside-induced nephrosis (PAN) rats, a well-established model of podocyte injury and human nephrotic syndrome. PAN was induced using a single intraperitoneal injection. Nicorandil was administered orally at 30 mg/kg/day. We found that proteinuria and hypoalbuminemia in PAN rats were significantly ameliorated following nicorandil treatment. Immunostaining and ultrastructural analysis under electron microscopy demonstrated that podocyte injury in PAN rats showed a significant partial attenuation following nicorandil treatment. Nicorandil ameliorated the increase in the oxidative stress markers nitrotyrosine and 8-hydroxy-2-deoxyguanosine in glomeruli. Conversely, nicorandil prevented the decrease in levels of the antioxidant enzyme manganese superoxide dismutase in PAN rats. We found that mitochondrial Ca<sup>2+</sup> uniporter levels in glomeruli were higher in PAN rats than in control rats, and this increase was significantly attenuated by nicorandil. We conclude that stimulation of MitK<sub>ATP</sub> by nicorandil reduces proteinuria by attenuating podocyte injury in PAN nephrosis, which restores mitochondrial antioxidative capacity, possibly through mitochondrial Ca<sup>2+</sup> uniporter modulation. These data indicate that MitK<sub>ATP</sub> may represent a novel target for podocyte injury and nephrotic syndrome.<b>NEW & NOTEWORTHY</b> Our findings suggest that the mitochondrial Ca<sup>2+</sup> uniporter may be an upstream regulator of manganese superoxide dismutase and indicate a biochemical basis for the interaction between the ATP-sensitive K<sup>+</sup> channel and Ca<sup>2+</sup> signaling. We believe that our study makes a significant contribution to the literature because our results indicate that the ATP-sensitive K<sup>+</sup> channel may be a potential therapeutic target for podocyte injury and nephrotic syndrome.</p>","PeriodicalId":7588,"journal":{"name":"American Journal of Physiology-renal Physiology","volume":"324 2","pages":"F168-F178"},"PeriodicalIF":3.7000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9844977/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Physiology-renal Physiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1152/ajprenal.00144.2022","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PHYSIOLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
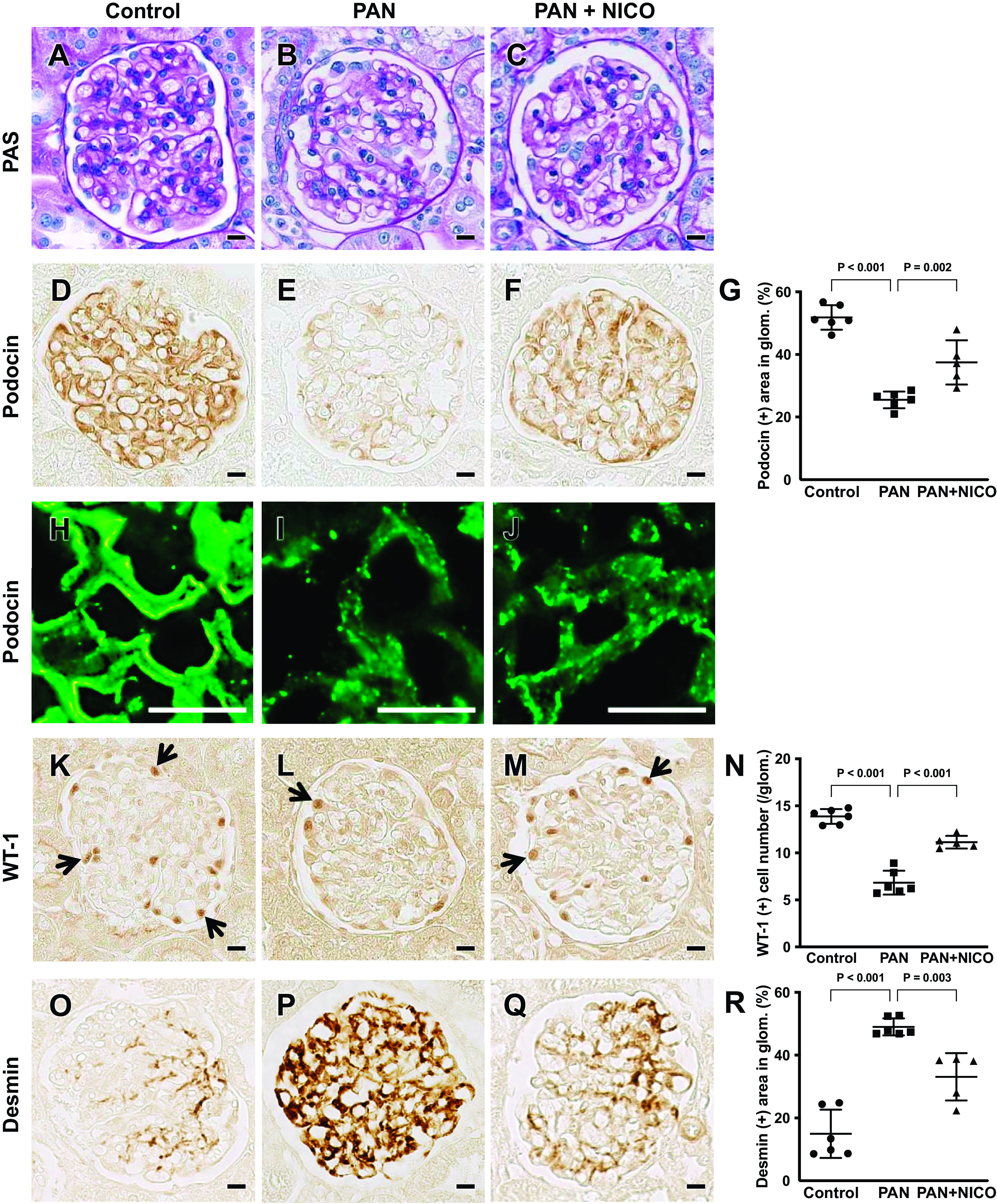
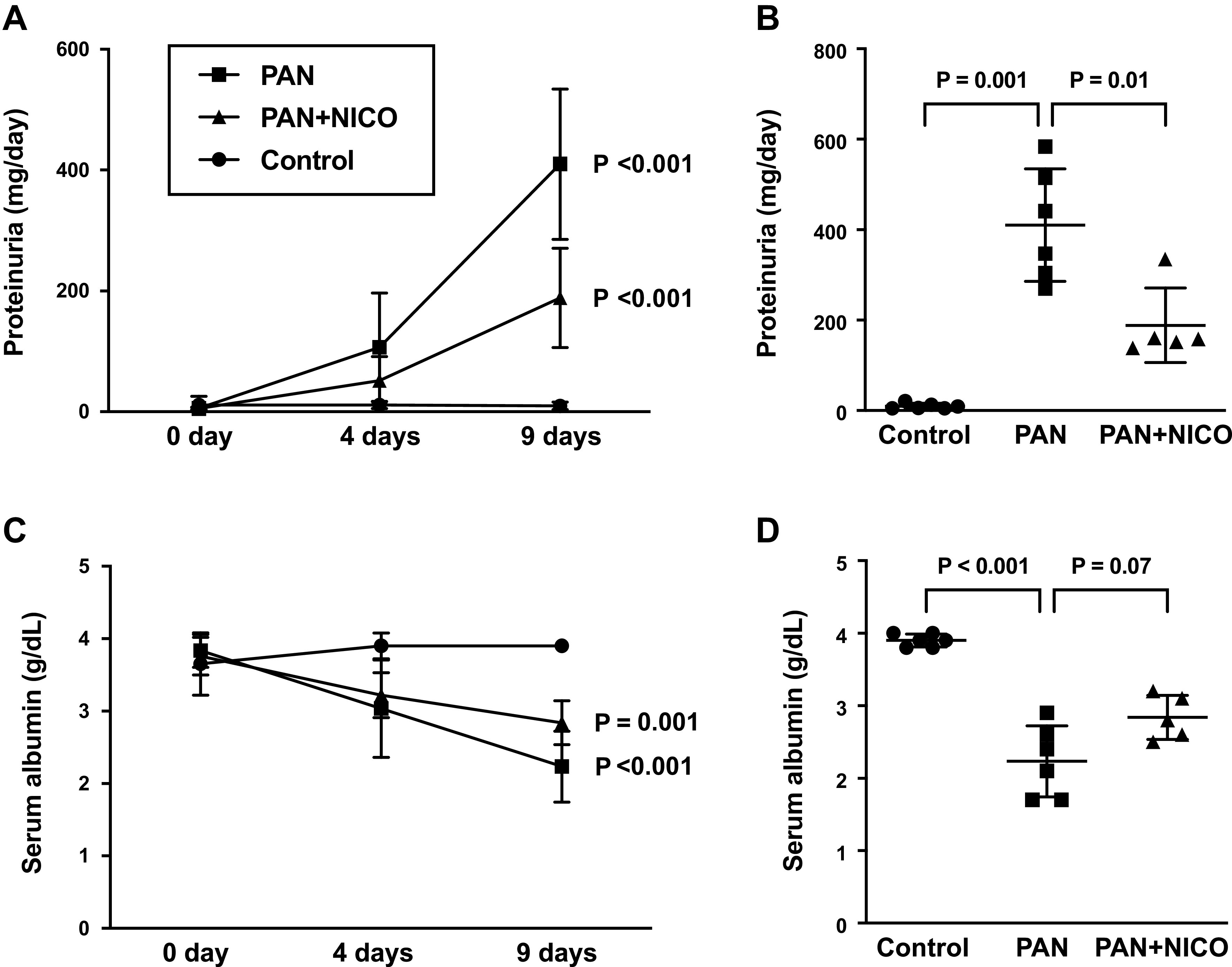
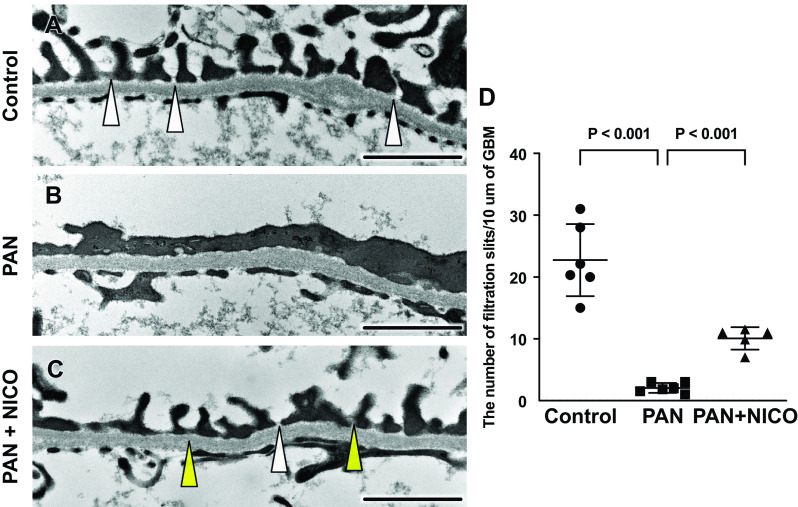
Nephrotic syndrome, characterized by proteinuria and hypoalbuminemia, results from the dysregulation of glomerular podocytes and is a significant cause of end-stage kidney disease. Patients with idiopathic nephrotic syndrome are generally treated with immunosuppressive agents; however, these agents produce various adverse effects. Previously, we reported the renoprotective effects of a stimulator of the mitochondrial ATP-dependent K+ channel (MitKATP), nicorandil, in a remnant kidney model. Nonetheless, the cellular targets of these effects remain unknown. Here, we examined the effect of nicorandil on puromycin aminonucleoside-induced nephrosis (PAN) rats, a well-established model of podocyte injury and human nephrotic syndrome. PAN was induced using a single intraperitoneal injection. Nicorandil was administered orally at 30 mg/kg/day. We found that proteinuria and hypoalbuminemia in PAN rats were significantly ameliorated following nicorandil treatment. Immunostaining and ultrastructural analysis under electron microscopy demonstrated that podocyte injury in PAN rats showed a significant partial attenuation following nicorandil treatment. Nicorandil ameliorated the increase in the oxidative stress markers nitrotyrosine and 8-hydroxy-2-deoxyguanosine in glomeruli. Conversely, nicorandil prevented the decrease in levels of the antioxidant enzyme manganese superoxide dismutase in PAN rats. We found that mitochondrial Ca2+ uniporter levels in glomeruli were higher in PAN rats than in control rats, and this increase was significantly attenuated by nicorandil. We conclude that stimulation of MitKATP by nicorandil reduces proteinuria by attenuating podocyte injury in PAN nephrosis, which restores mitochondrial antioxidative capacity, possibly through mitochondrial Ca2+ uniporter modulation. These data indicate that MitKATP may represent a novel target for podocyte injury and nephrotic syndrome.NEW & NOTEWORTHY Our findings suggest that the mitochondrial Ca2+ uniporter may be an upstream regulator of manganese superoxide dismutase and indicate a biochemical basis for the interaction between the ATP-sensitive K+ channel and Ca2+ signaling. We believe that our study makes a significant contribution to the literature because our results indicate that the ATP-sensitive K+ channel may be a potential therapeutic target for podocyte injury and nephrotic syndrome.
期刊介绍:
The American Journal of Physiology - Renal Physiology publishes original manuscripts on timely topics in both basic science and clinical research. Published articles address a broad range of subjects relating to the kidney and urinary tract, and may involve human or animal models, individual cell types, and isolated membrane systems. Also covered are the pathophysiological basis of renal disease processes, regulation of body fluids, and clinical research that provides mechanistic insights. Studies of renal function may be conducted using a wide range of approaches, such as biochemistry, immunology, genetics, mathematical modeling, molecular biology, as well as physiological and clinical methodologies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


