{"title":"<i>In-silico</i> Structural Modeling of Human Immunodeficiency Virus Proteins.","authors":"Amir Elalouf","doi":"10.1177/11795972231154402","DOIUrl":null,"url":null,"abstract":"<p><p>Human immunodeficiency virus (HIV) is an infectious virus that depletes the CD4<sup>+</sup> <i>T</i> lymphocytes of the immune system and causes a chronic life-treating disease-acquired immunodeficiency syndrome (AIDS). The HIV genome encodes different structural and accessory proteins involved in viral entry and life cycle. Determining the 3D structure of HIV proteins is essential for new target position finding, structure-based drug designing, and future planning for computational and laboratory experimentations. Hence, the study aims to predict the 3D structures of all the HIV structural and accessory proteins using computational homology modeling to understand better the structural basis of HIV proteins interacting with host cells and viral replication. The sequences of HIV capsid, matrix, nucleocapsid, p6, reverse transcriptase, invertase, protease, gp120, gp41, virus protein r, viral infectivity factor, virus protein unique, RNA splicing regulator, transactivator protein, negative regulating factor, and virus protein x proteins were retrieved from UniProt. The primary and secondary structures of HIV proteins were predicted by Expasy ProtParam and SOPMA web servers. For the homology modeling, the MODELLER predicted the 3D structures of HIV proteins using templates. Then, the modeled structures were validated by the Ramachandran plot, local and global quality estimation scores, QMEAN scores, and <i>Z</i>-scores. Most of the amino acid residues of HIV proteins were present in the most favored and generously allowed regions in the Ramachandran plots. The local and global quality scores and <i>Z</i>-scores of the HIV proteins confirmed the good quality of modeled structures. The 3D modeled structures of HIV proteins might help further investigate the possible treatment.</p>","PeriodicalId":42484,"journal":{"name":"Biomedical Engineering and Computational Biology","volume":"14 ","pages":"11795972231154402"},"PeriodicalIF":3.1000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8d/21/10.1177_11795972231154402.PMC9936402.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomedical Engineering and Computational Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11795972231154402","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENGINEERING, BIOMEDICAL","Score":null,"Total":0}
引用次数: 0
Abstract
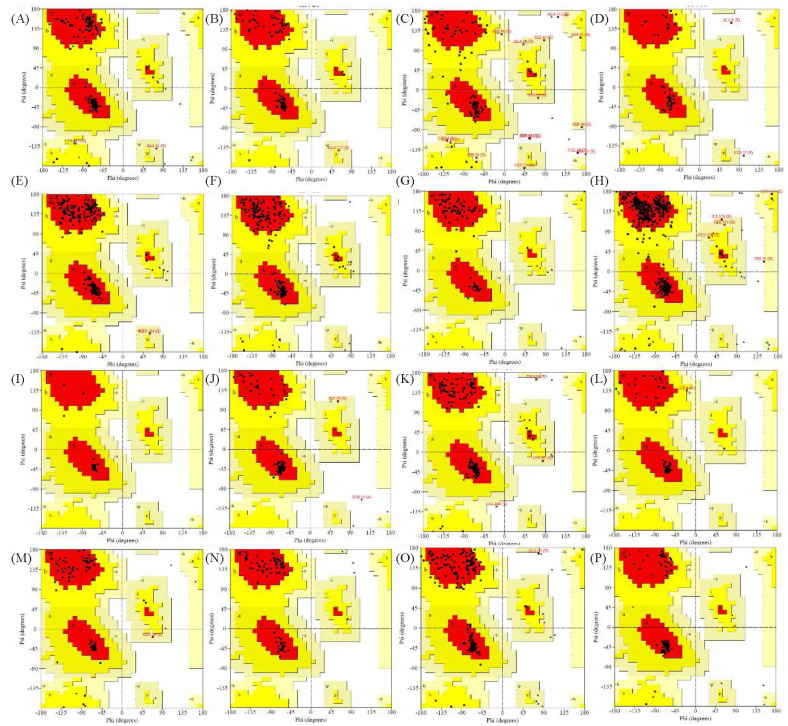
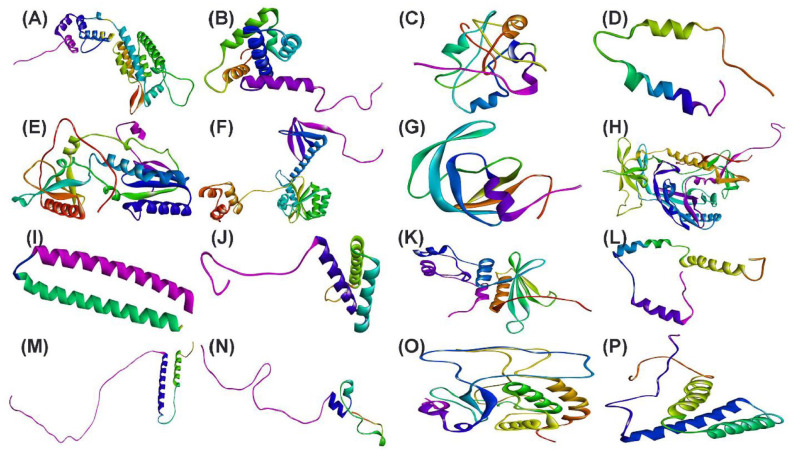
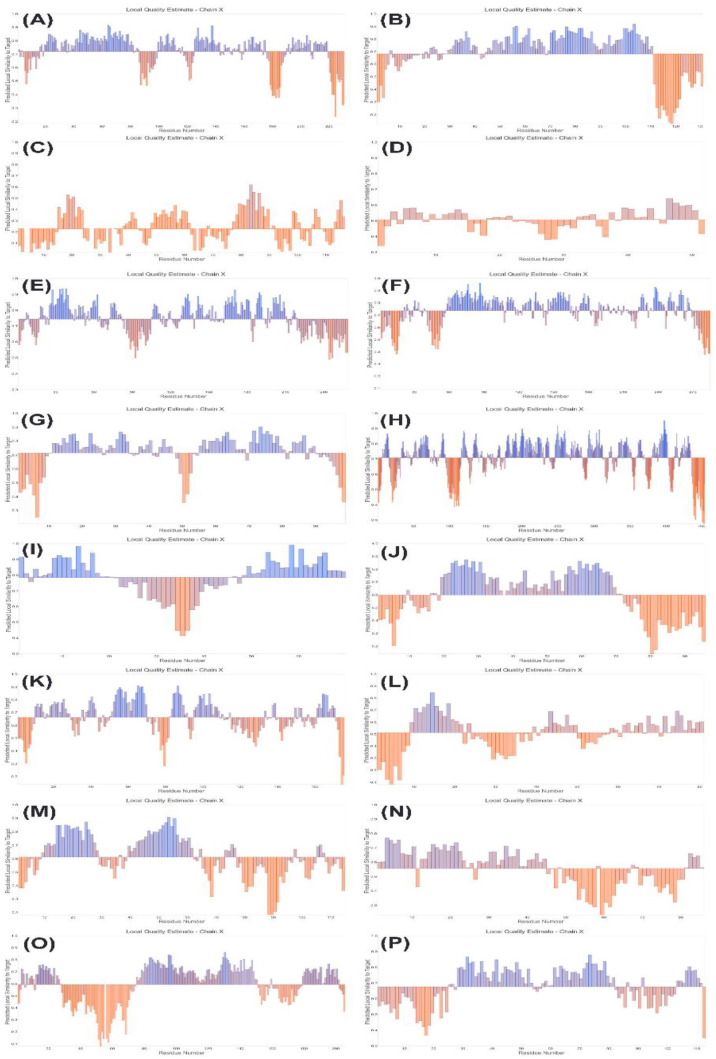
Human immunodeficiency virus (HIV) is an infectious virus that depletes the CD4+T lymphocytes of the immune system and causes a chronic life-treating disease-acquired immunodeficiency syndrome (AIDS). The HIV genome encodes different structural and accessory proteins involved in viral entry and life cycle. Determining the 3D structure of HIV proteins is essential for new target position finding, structure-based drug designing, and future planning for computational and laboratory experimentations. Hence, the study aims to predict the 3D structures of all the HIV structural and accessory proteins using computational homology modeling to understand better the structural basis of HIV proteins interacting with host cells and viral replication. The sequences of HIV capsid, matrix, nucleocapsid, p6, reverse transcriptase, invertase, protease, gp120, gp41, virus protein r, viral infectivity factor, virus protein unique, RNA splicing regulator, transactivator protein, negative regulating factor, and virus protein x proteins were retrieved from UniProt. The primary and secondary structures of HIV proteins were predicted by Expasy ProtParam and SOPMA web servers. For the homology modeling, the MODELLER predicted the 3D structures of HIV proteins using templates. Then, the modeled structures were validated by the Ramachandran plot, local and global quality estimation scores, QMEAN scores, and Z-scores. Most of the amino acid residues of HIV proteins were present in the most favored and generously allowed regions in the Ramachandran plots. The local and global quality scores and Z-scores of the HIV proteins confirmed the good quality of modeled structures. The 3D modeled structures of HIV proteins might help further investigate the possible treatment.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


