Arkaprava Banerjee, Agnieszka Gajewicz-Skretna, K Roy
{"title":"A machine learning q-RASPR approach for efficient predictions of the specific surface area of perovskites.","authors":"Arkaprava Banerjee, Agnieszka Gajewicz-Skretna, K Roy","doi":"10.1002/minf.202200261","DOIUrl":null,"url":null,"abstract":"<p><p>In this study, the specific surface area of various perovskites was modeled using a novel quantitative read-across structure-property relationship (q-RASPR) approach, which clubs both Read-Across (RA) and quantitative structure-property relationship (QSPR) together. After optimization of the hyper-parameters, certain similarity-based error measures for each query compound were obtained. Clubbing some of these error-based measures with the previously selected features along with the Read-Across prediction function, a number of machine learning models were developed using Partial Least Squares (PLS), Ridge Regression (RR), Linear Support Vector Regression (LSVR), Random Forest (RF) regression, Gradient Boost (GBoost), Adaptive Boosting (Adaboost), Multiple Layer Perceptron (MLP) regression and k-Nearest Neighbor (kNN) regression. Based on the repeated cross-validation as well as external prediction quality and interpretability, the PLS model (n<sub>Training</sub> = 38, n<sub>Test</sub> = 12, <math> <semantics><msubsup><mi>R</mi> <mrow><mi>T</mi> <mi>r</mi> <mi>a</mi> <mi>i</mi> <mi>n</mi></mrow> <mn>2</mn></msubsup> <annotation>${{R}_{Train}^{2}}$</annotation> </semantics> </math> =0.737, <math> <semantics> <mrow><msubsup><mi>Q</mi> <mrow><mi>L</mi> <mi>O</mi> <mi>O</mi></mrow> <mn>2</mn></msubsup> <mo>=</mo> <mn>0</mn> <mo>.</mo> <mn>637</mn> <mo>,</mo> <mspace></mspace> <msubsup><mi>R</mi> <mrow><mi>T</mi> <mi>e</mi> <mi>s</mi> <mi>t</mi></mrow> <mn>2</mn></msubsup> <mo>=</mo> <mn>0</mn> <mo>.</mo> <mn>898</mn> <mo>,</mo> <mspace></mspace> <mspace></mspace> <msubsup><mi>Q</mi> <mrow><mi>F</mi> <mn>1</mn> <mfenced><mi>T</mi> <mi>e</mi> <mi>s</mi> <mi>t</mi></mfenced> </mrow> <mn>2</mn></msubsup> <mrow><mo>=</mo> <mn>0</mn> <mo>.</mo> <mn>901</mn> <mo>)</mo></mrow> </mrow> <annotation>${{Q}_{LOO}^{2}=0.637,\\ {R}_{Test}^{2}=0.898,{\\rm \\ }\\ {Q}_{F1\\left(Test\\right)}^{2}=0.901)}$</annotation> </semantics> </math> was selected as the best predictor which underscored the previously reported results. The finally selected model should efficiently predict specific surface areas of other perovskites for their use in photocatalysis. The new q-RASPR method also appears promising for the prediction of several other property endpoints of interest in materials science.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"42 4","pages":"e2200261"},"PeriodicalIF":2.8000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200261","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 6
Abstract
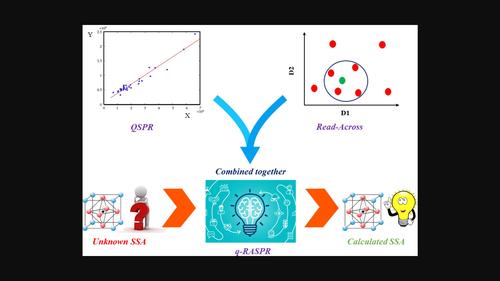
In this study, the specific surface area of various perovskites was modeled using a novel quantitative read-across structure-property relationship (q-RASPR) approach, which clubs both Read-Across (RA) and quantitative structure-property relationship (QSPR) together. After optimization of the hyper-parameters, certain similarity-based error measures for each query compound were obtained. Clubbing some of these error-based measures with the previously selected features along with the Read-Across prediction function, a number of machine learning models were developed using Partial Least Squares (PLS), Ridge Regression (RR), Linear Support Vector Regression (LSVR), Random Forest (RF) regression, Gradient Boost (GBoost), Adaptive Boosting (Adaboost), Multiple Layer Perceptron (MLP) regression and k-Nearest Neighbor (kNN) regression. Based on the repeated cross-validation as well as external prediction quality and interpretability, the PLS model (nTraining = 38, nTest = 12, =0.737, was selected as the best predictor which underscored the previously reported results. The finally selected model should efficiently predict specific surface areas of other perovskites for their use in photocatalysis. The new q-RASPR method also appears promising for the prediction of several other property endpoints of interest in materials science.
在本研究中,使用一种新的定量跨读结构-性质关系(q-RASPR)方法对各种钙钛矿的比表面积进行了建模,该方法将跨读(RA)和定量结构-性质关系(QSPR)结合在一起。通过对超参数的优化,得到了基于相似性的误差度量。将其中一些基于误差的度量与先前选择的特征以及Read-Across预测函数结合起来,使用偏最小二乘(PLS)、岭回归(RR)、线性支持向量回归(LSVR)、随机森林(RF)回归、梯度增强(GBoost)、自适应增强(Adaboost)、多层感知器(MLP)回归和k-最近邻(kNN)回归开发了许多机器学习模型。基于重复交叉验证以及外部预测质量和可解释性,PLS模型(nTraining = 38, nTest = 12, R T R ain 2 ${{R}_{Train}^{2}}$ =0.737, Q L O O 2 =0。637 R T T s T 2 = 0。qf1t = 0。901) ${{Q}_{LOO}^{2}=0.637,\ {R}_{Test}^{2}=0.898,{\rm \}\ {Q}_{F1\left(Test\right)}^{2}=0.901)}$被选为最佳预测因子,强调了先前报道的结果。最后选择的模型应该能够有效地预测其他钙钛矿在光催化中的比表面积。新的q-RASPR方法似乎也有希望预测材料科学中其他几个感兴趣的属性端点。
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


