Kushagra Kashyap, Lalita Panigrahi, Shakil Ahmed, Mohammad Siddiqi
{"title":"Artificial neural network models driven novel virtual screening workflow for the identification and biological evaluation of BACE1 inhibitors.","authors":"Kushagra Kashyap, Lalita Panigrahi, Shakil Ahmed, Mohammad Siddiqi","doi":"10.1002/minf.202200113","DOIUrl":null,"url":null,"abstract":"<p><p>Beta-site amyloid-β precursor protein-cleaving enzyme 1 (BACE1) is a transmembrane aspartic protease and has shown potential as a possible therapeutic target for Alzheimer's disease. This aggravating disease involves the aberrant production of β amyloid plaques by BACE1 which catalyzes the rate-limiting step by cleaving the amyloid precursor protein (APP), generating the neurotoxic amyloid β protein that aggregates to form plaques leading to neurodegeneration. Therefore, it is indispensable to inhibit BACE1, thus modulating the APP processing. In this study, we present a workflow that utilizes a multi-stage virtual screening protocol for identifying potential BACE1 inhibitors by employing multiple artificial neural network-based models. Collectively, all the hyperparameter tuned models were assigned a task to virtually screen Maybridge library, thus yielding a consensus of 41 hits. The majority of these hits exhibited optimal pharmacokinetic properties confirmed by high central nervous system multiparameter optimization (CNS-MPO) scores. Further shortlisting of 8 compounds by molecular docking into the active site of BACE1 and their subsequent in-vitro evaluation identified 4 compounds as potent BACE1 inhibitors with IC50 values falling in the range 0.028-0.052 μM and can be further optimized with medicinal chemistry efforts to improve their activity.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"42 3","pages":"e2200113"},"PeriodicalIF":2.8000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200113","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 2
Abstract
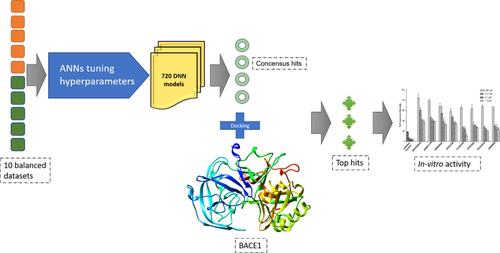
Beta-site amyloid-β precursor protein-cleaving enzyme 1 (BACE1) is a transmembrane aspartic protease and has shown potential as a possible therapeutic target for Alzheimer's disease. This aggravating disease involves the aberrant production of β amyloid plaques by BACE1 which catalyzes the rate-limiting step by cleaving the amyloid precursor protein (APP), generating the neurotoxic amyloid β protein that aggregates to form plaques leading to neurodegeneration. Therefore, it is indispensable to inhibit BACE1, thus modulating the APP processing. In this study, we present a workflow that utilizes a multi-stage virtual screening protocol for identifying potential BACE1 inhibitors by employing multiple artificial neural network-based models. Collectively, all the hyperparameter tuned models were assigned a task to virtually screen Maybridge library, thus yielding a consensus of 41 hits. The majority of these hits exhibited optimal pharmacokinetic properties confirmed by high central nervous system multiparameter optimization (CNS-MPO) scores. Further shortlisting of 8 compounds by molecular docking into the active site of BACE1 and their subsequent in-vitro evaluation identified 4 compounds as potent BACE1 inhibitors with IC50 values falling in the range 0.028-0.052 μM and can be further optimized with medicinal chemistry efforts to improve their activity.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


