Ajay H Bhandarwar, Amarjeet E Tandur, Keerthika Reddy Rachapalli, Amol Wagh, Abhijit Shah, Nikhil Dhimole
{"title":"Minimally Invasive Management of Hemorrhagic Pheochromocytoma-A Rare Case Report.","authors":"Ajay H Bhandarwar, Amarjeet E Tandur, Keerthika Reddy Rachapalli, Amol Wagh, Abhijit Shah, Nikhil Dhimole","doi":"10.1055/s-0043-1762554","DOIUrl":null,"url":null,"abstract":"<p><p>Pheochromocytoma is a rare catecholamine-secreting tumor derived from chromaffin cells. The diagnosis is usually suggested by its classic history, presence of a strong family history, or discovery of an incidental mass on imaging in an asymptomatic patient. Hemorrhage into an occult pheochromocytoma is a rare complication with ∼1 to 2 per 100,000 individuals diagnosed annually. We report a case of a 29-year-old woman, who presented with abdominal pain (with no other significant history) due to a right hemorrhagic pheochromocytoma. Computed tomographic imaging and magnetic resonance imaging revealed the source of retroperitoneal hemorrhage as the right adrenal mass. They lacked the typical features of a pheochromocytoma which was eventually proven by the biochemical tests. The patient underwent preoperative stabilization with α and β adrenergic receptor blockers for 7 days following which laparoscopic adrenalectomy was performed successfully with an uneventful postoperative period. This is the eighth reported case in literature managed laparoscopically. Histopathology confirmed it as pheochromocytoma. The treacherous and deceptive nature of pheochromocytomas and its hemorrhage make it crucial to detect and treat it promptly; otherwise, it will almost certainly be fatal from cardiovascular complications or metastasis.</p>","PeriodicalId":0,"journal":{"name":"","volume":null,"pages":null},"PeriodicalIF":0.0,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10040260/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1055/s-0043-1762554","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
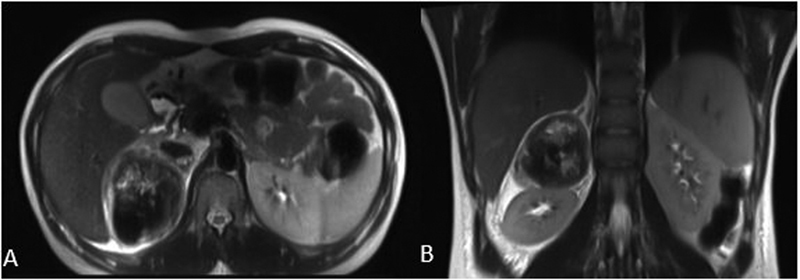
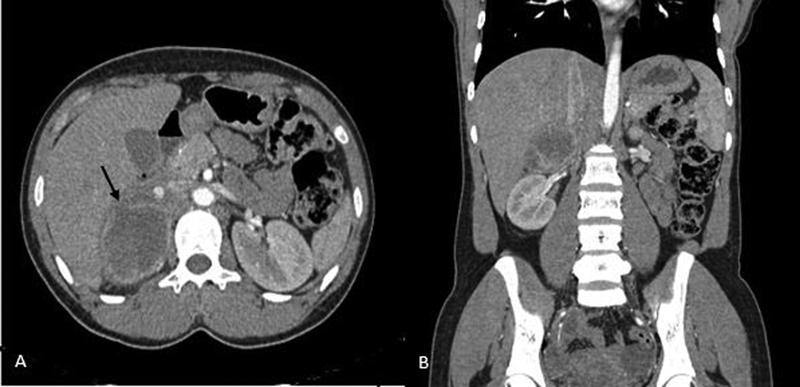
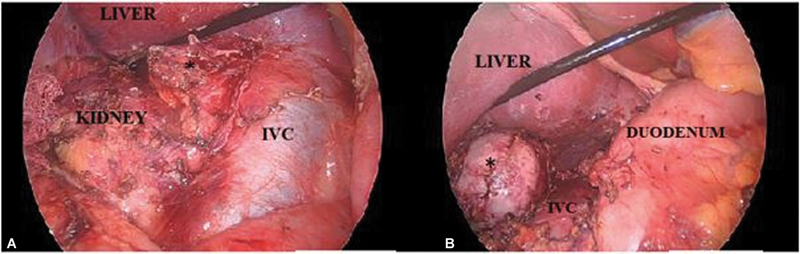
Pheochromocytoma is a rare catecholamine-secreting tumor derived from chromaffin cells. The diagnosis is usually suggested by its classic history, presence of a strong family history, or discovery of an incidental mass on imaging in an asymptomatic patient. Hemorrhage into an occult pheochromocytoma is a rare complication with ∼1 to 2 per 100,000 individuals diagnosed annually. We report a case of a 29-year-old woman, who presented with abdominal pain (with no other significant history) due to a right hemorrhagic pheochromocytoma. Computed tomographic imaging and magnetic resonance imaging revealed the source of retroperitoneal hemorrhage as the right adrenal mass. They lacked the typical features of a pheochromocytoma which was eventually proven by the biochemical tests. The patient underwent preoperative stabilization with α and β adrenergic receptor blockers for 7 days following which laparoscopic adrenalectomy was performed successfully with an uneventful postoperative period. This is the eighth reported case in literature managed laparoscopically. Histopathology confirmed it as pheochromocytoma. The treacherous and deceptive nature of pheochromocytomas and its hemorrhage make it crucial to detect and treat it promptly; otherwise, it will almost certainly be fatal from cardiovascular complications or metastasis.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


