{"title":"The Spectrum of Familial Pituitary Neuroendocrine Tumors.","authors":"Eleni Armeni, Ashley Grossman","doi":"10.1007/s12022-022-09742-0","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary pituitary tumorigenesis is seen in a relatively small proportion (around 5%) of patients with pituitary neuroendocrine tumors (PitNETs). The aim of the current review is to describe the main clinical and molecular features of such pituitary tumors associated with hereditary or familial characteristics, many of which have now been genetically identified. The genetic patterns of inheritance are classified into isolated familial PitNETs and the syndromic tumors. In general, the established genetic causes of familial tumorigenesis tend to present at a younger age, often pursue a more aggressive course, and are more frequently associated with growth hormone hypersecretion compared to sporadic tumors. The mostly studied molecular pathways implicated are the protein kinase A and phosphatidyl-inositol pathways, which are in the main related to mutations in the syndromes of familial isolated pituitary adenoma (FIPA), Carney complex syndrome, and X-linked acrogigantism. Another well-documented mechanism consists of the regulation of p27 or p21 proteins, with further acceleration of the pituitary cell cycle through the check points G1/S and M/G1, mostly documented in multiple endocrine neoplasia type 4. In conclusion, PitNETs may occur in relation to well-established familial germline mutations which may determine the clinical phenotype and the response to treatment, and may require family screening.</p>","PeriodicalId":55167,"journal":{"name":"Endocrine Pathology","volume":"34 1","pages":"57-78"},"PeriodicalIF":11.3000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine Pathology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12022-022-09742-0","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 2
Abstract
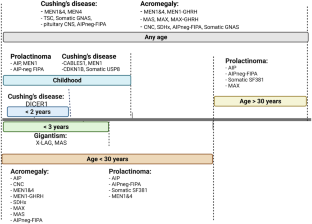
Hereditary pituitary tumorigenesis is seen in a relatively small proportion (around 5%) of patients with pituitary neuroendocrine tumors (PitNETs). The aim of the current review is to describe the main clinical and molecular features of such pituitary tumors associated with hereditary or familial characteristics, many of which have now been genetically identified. The genetic patterns of inheritance are classified into isolated familial PitNETs and the syndromic tumors. In general, the established genetic causes of familial tumorigenesis tend to present at a younger age, often pursue a more aggressive course, and are more frequently associated with growth hormone hypersecretion compared to sporadic tumors. The mostly studied molecular pathways implicated are the protein kinase A and phosphatidyl-inositol pathways, which are in the main related to mutations in the syndromes of familial isolated pituitary adenoma (FIPA), Carney complex syndrome, and X-linked acrogigantism. Another well-documented mechanism consists of the regulation of p27 or p21 proteins, with further acceleration of the pituitary cell cycle through the check points G1/S and M/G1, mostly documented in multiple endocrine neoplasia type 4. In conclusion, PitNETs may occur in relation to well-established familial germline mutations which may determine the clinical phenotype and the response to treatment, and may require family screening.
期刊介绍:
Endocrine Pathology publishes original articles on clinical and basic aspects of endocrine disorders. Work with animals or in vitro techniques is acceptable if it is relevant to human normal or abnormal endocrinology. Manuscripts will be considered for publication in the form of original articles, case reports, clinical case presentations, reviews, and descriptions of techniques. Submission of a paper implies that it reports unpublished work, except in abstract form, and is not being submitted simultaneously to another publication. Accepted manuscripts become the sole property of Endocrine Pathology and may not be published elsewhere without written consent from the publisher. All articles are subject to review by experienced referees. The Editors and Editorial Board judge manuscripts suitable for publication, and decisions by the Editors are final.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


