Tristan Hoellinger, Camille Mestre, Hugues Aschard, Wilfried Le Goff, Sylvain Foissac, Thomas Faraut, Sarah Djebali
{"title":"Enhancer/gene relationships: Need for more reliable genome-wide reference sets.","authors":"Tristan Hoellinger, Camille Mestre, Hugues Aschard, Wilfried Le Goff, Sylvain Foissac, Thomas Faraut, Sarah Djebali","doi":"10.3389/fbinf.2023.1092853","DOIUrl":null,"url":null,"abstract":"<p><p>Differences in cells' functions arise from differential activity of regulatory elements, including enhancers. Enhancers are cis-regulatory elements that cooperate with promoters through transcription factors to activate the expression of one or several genes by getting physically close to them in the 3D space of the nucleus. There is increasing evidence that genetic variants associated with common diseases are enriched in enhancers active in cell types relevant to these diseases. Identifying the enhancers associated with genes and conversely, the sets of genes activated by each enhancer (the so-called enhancer/gene or E/G relationships) across cell types, can help understanding the genetic mechanisms underlying human diseases. There are three broad approaches for the genome-wide identification of E/G relationships in a cell type: 1) genetic link methods or eQTL, 2) functional link methods based on 1D functional data such as open chromatin, histone mark or gene expression and 3) spatial link methods based on 3D data such as HiC. Since 1) and 3) are costly, the current strategy is to develop functional link methods and to use data from 1) and 3) as reference to evaluate them. However, there is still no consensus on the best functional link method to date, and method comparison remain seldom. Here, we compared the relative performances of three recent methods for the identification of enhancer-gene links, TargetFinder, Average-Rank, and the ABC model, using the three latest benchmarks from the field: a reference that combines 3D and eQTL data, called BENGI, and two genetic screening references, called CRiFF and CRiSPRi. Overall, none of the three methods performed best on the three references. CRiFF and CRISPRi reference sets are likely more reliable, but CRiFF is not genome-wide and CRiFF and CRISPRi are mostly available on the K562 cancer cell line. The BENGI reference set is genome-wide but likely contains many false positives. This study therefore calls for new reliable and genome-wide E/G reference data rather than new functional link E/G identification methods.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"3 ","pages":"1092853"},"PeriodicalIF":2.8000,"publicationDate":"2023-02-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9999192/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2023.1092853","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
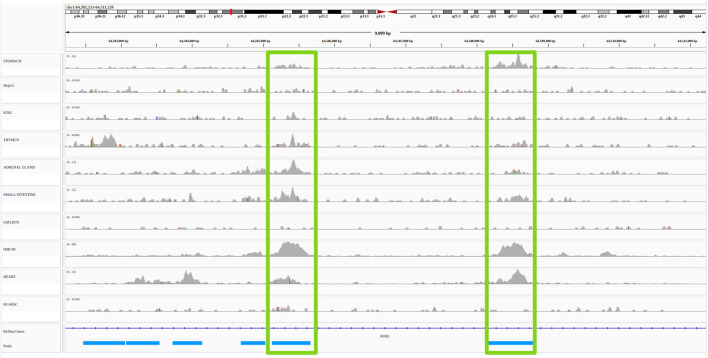
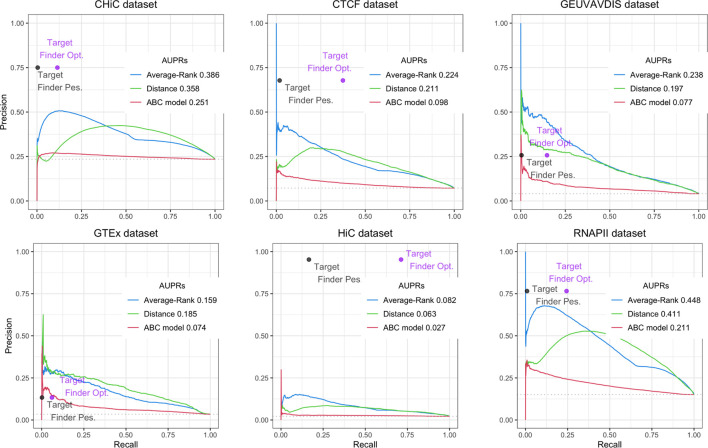
Differences in cells' functions arise from differential activity of regulatory elements, including enhancers. Enhancers are cis-regulatory elements that cooperate with promoters through transcription factors to activate the expression of one or several genes by getting physically close to them in the 3D space of the nucleus. There is increasing evidence that genetic variants associated with common diseases are enriched in enhancers active in cell types relevant to these diseases. Identifying the enhancers associated with genes and conversely, the sets of genes activated by each enhancer (the so-called enhancer/gene or E/G relationships) across cell types, can help understanding the genetic mechanisms underlying human diseases. There are three broad approaches for the genome-wide identification of E/G relationships in a cell type: 1) genetic link methods or eQTL, 2) functional link methods based on 1D functional data such as open chromatin, histone mark or gene expression and 3) spatial link methods based on 3D data such as HiC. Since 1) and 3) are costly, the current strategy is to develop functional link methods and to use data from 1) and 3) as reference to evaluate them. However, there is still no consensus on the best functional link method to date, and method comparison remain seldom. Here, we compared the relative performances of three recent methods for the identification of enhancer-gene links, TargetFinder, Average-Rank, and the ABC model, using the three latest benchmarks from the field: a reference that combines 3D and eQTL data, called BENGI, and two genetic screening references, called CRiFF and CRiSPRi. Overall, none of the three methods performed best on the three references. CRiFF and CRISPRi reference sets are likely more reliable, but CRiFF is not genome-wide and CRiFF and CRISPRi are mostly available on the K562 cancer cell line. The BENGI reference set is genome-wide but likely contains many false positives. This study therefore calls for new reliable and genome-wide E/G reference data rather than new functional link E/G identification methods.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


