{"title":"Modeling Nanostructure in Graphene Oxide: Inhomogeneity and the Percolation Threshold","authors":"Robert C. Sinclair*, Peter V. Coveney","doi":"10.1021/acs.jcim.9b00114","DOIUrl":null,"url":null,"abstract":"<p >Graphene oxide (GO) is an amorphous 2D material, which has found widespread use in the fields of chemistry, physics, and materials science due to its similarity to graphene with the benefit of being far easier to synthesize and process. However, the standard of GO characterization is very poor because its structure is irregular, being sensitive to the preparation method, and it has a propensity to transform due to its reactive nature. Atomistic simulations of GO are common, but the nanostructure in these simulations is often based on little evidence or thought. We have written a computer program to generate graphene oxide nanostructures for general purpose atomistic simulation based on theoretical and experimental evidence. The structures generated offer a significant improvement to the current standard of randomly placed oxidized functional groups and successfully recreate the two-phase nature of oxidized and unoxidized graphene domains observed in microscopy experiments. Using this model, we reveal new features of GO structure and predict that a critical point in the oxidation reaction exists as the oxidized region reaches a percolation threshold. Even by a conservative estimate, we show that, if the carbon to oxygen ratio is kept above 6, a continuous aromatic network will remain, preserving many of graphene’s desirable properties, irrespective of the oxidation method or the size distribution of graphene sheets. This is an experimentally achievable degree of oxidation and should aid better GO synthesis for many applications.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"59 6","pages":"2741–2745"},"PeriodicalIF":5.3000,"publicationDate":"2019-04-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1021/acs.jcim.9b00114","citationCount":"44","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.9b00114","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 44
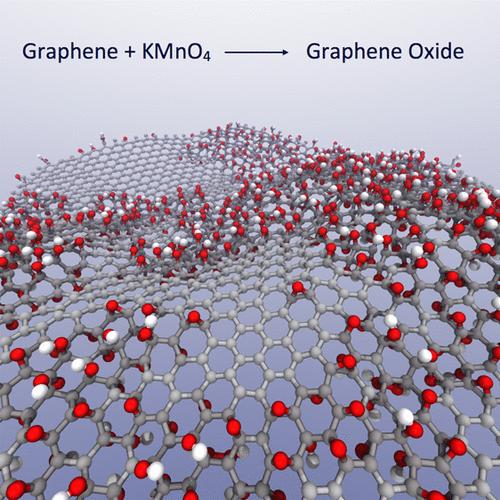
Abstract
Graphene oxide (GO) is an amorphous 2D material, which has found widespread use in the fields of chemistry, physics, and materials science due to its similarity to graphene with the benefit of being far easier to synthesize and process. However, the standard of GO characterization is very poor because its structure is irregular, being sensitive to the preparation method, and it has a propensity to transform due to its reactive nature. Atomistic simulations of GO are common, but the nanostructure in these simulations is often based on little evidence or thought. We have written a computer program to generate graphene oxide nanostructures for general purpose atomistic simulation based on theoretical and experimental evidence. The structures generated offer a significant improvement to the current standard of randomly placed oxidized functional groups and successfully recreate the two-phase nature of oxidized and unoxidized graphene domains observed in microscopy experiments. Using this model, we reveal new features of GO structure and predict that a critical point in the oxidation reaction exists as the oxidized region reaches a percolation threshold. Even by a conservative estimate, we show that, if the carbon to oxygen ratio is kept above 6, a continuous aromatic network will remain, preserving many of graphene’s desirable properties, irrespective of the oxidation method or the size distribution of graphene sheets. This is an experimentally achievable degree of oxidation and should aid better GO synthesis for many applications.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


