Efficient Bayesian inference of instantaneous reproduction numbers at fine spatial scales, with an application to mapping and nowcasting the Covid-19 epidemic in British local authorities
IF 1.5 3区 数学Q2 SOCIAL SCIENCES, MATHEMATICAL METHODS
Yee Whye Teh, Bryn Elesedy, Bobby He, Michael Hutchinson, Sheheryar Zaidi, Avishkar Bhoopchand, Ulrich Paquet, Nenad Tomasev, Jonathan Read, Peter J. Diggle
{"title":"Efficient Bayesian inference of instantaneous reproduction numbers at fine spatial scales, with an application to mapping and nowcasting the Covid-19 epidemic in British local authorities","authors":"Yee Whye Teh, Bryn Elesedy, Bobby He, Michael Hutchinson, Sheheryar Zaidi, Avishkar Bhoopchand, Ulrich Paquet, Nenad Tomasev, Jonathan Read, Peter J. Diggle","doi":"10.1111/rssa.12971","DOIUrl":null,"url":null,"abstract":"<p>The spatio-temporal pattern of Covid-19 infections, as for most infectious disease epidemics, is highly heterogenous as a consequence of local variations in risk factors and exposures. Consequently, the widely quoted national-level estimates of reproduction numbers are of limited value in guiding local interventions and monitoring their effectiveness. It is crucial for national and local policy-makers, and for health protection teams, that accurate, well-calibrated and timely predictions of Covid-19 incidences and transmission rates are available at fine spatial scales. Obtaining such estimates is challenging, not least due to the prevalence of asymptomatic Covid-19 transmissions, as well as difficulties of obtaining high-resolution and high-frequency data. In addition, low case counts at a local level further confounds the inference for Covid-19 transmission rates, adding unwelcome uncertainty.</p><p>In this paper we develop a hierarchical Bayesian method for inference of transmission rates at fine spatial scales. Our model incorporates both temporal and spatial dependencies of local transmission rates in order to share statistical strength and reduce uncertainty. It also incorporates information about population flows to model potential transmissions across local areas. A simple approach to posterior simulation quickly becomes computationally infeasible, which is problematic if the system is required to provide timely predictions. We describe how to make posterior simulation for the model efficient, so that we are able to provide daily updates on epidemic developments.</p><p>The results can be found at our web site https://localcovid.info, which is updated daily to display estimated instantaneous reproduction numbers and predicted case counts for the next weeks, across local authorities in Great Britain. The codebase updating the web site can be found at https://github.com/oxcsml/Rmap. We hope that our methodology and web site will be of interest to researchers, policy-makers and the public alike, to help identify upcoming local outbreaks and to aid in the containment of Covid-19 through both public health measures and personal decisions taken by the general public.</p><p>Our model is applied to publicly available daily counts of positive test results reported under the combined Pillars 1 (NHS and PHE) and 2 (commercial partners) of the UK's Covid-19 testing strategy.1 The data are available for 312 lower-tier local authorities (LTLAs) in England, 14 NHS Health Boards in Scotland (each covering multiple local authorities) and 22 unitary local authorities in Wales, for a total of <math>\n <semantics>\n <mrow>\n <mi>n</mi>\n <mo>=</mo>\n <mn>348</mn>\n </mrow>\n <annotation>$$ n=348 $$</annotation>\n </semantics></math> local areas. The data are daily counts of lab-confirmed (PCR swab) cases presented by specimen date, starting from 30 January 2020. The original data are from the respective national public health authorities of England2, Scotland3and Wales4and we access them through the DELVE Global Covid-19 Dataset5 (Bhoopchand et al., <span>2020</span>). Due to delays in processing tests, we ignore the last 7 days of case counts.</p><p>Our method is based on an approach to infectious disease modelling using discrete renewal processes. These have a long history, and have served as the basis for a number of recent studies estimating instantaneous reproduction numbers, (Cori et al., <span>2013</span>; Flaxman et al., <span>2020</span>; Fraser, <span>2007</span>; Wallinga & Teunis, <span>2004</span>). See Bhatt et al. (<span>2020</span>) and references therein for historical and mathematical background, as well as Gostic et al. (<span>2020</span>) for important practical considerations.</p><p>Following Flaxman et al. (<span>2020</span>), we model latent time series of incidence rates via renewal processes, and separate observations of reported cases using negative binomial distributions, to account for uncertainties in case reporting, outliers in case counts, and delays between infection and testing. We introduce a number of extensions and differences addressing issues that arise for applications to modelling epidemics at local authority level rather than regional or national levels. Firstly, we introduce dependencies between reproduction numbers across neighbouring localities, in order to smooth estimates of reproduction numbers and statistical strength across localities and time. We do this using a spatio-temporal Gaussian process (GP) prior for the log-transformed reproduction numbers. Secondly, we model transmissions across localities using a spatial meta-population model. Our meta-population model incorporates commuter flow data from the UK 2011 Census in order to capture stable patterns of heterogenous cross-infection rates among local authorities, linked to typical commuter patterns. Human mobility patterns may also reflect the introduction of non-pharmaceutical interventions (NPIs), though our model does not explicitly use real-time mobility data so cannot estimate the direct or indirect effects of NPIs.</p><p>The model is implemented in the Stan probabilistic programming language (Carpenter et al., <span>2017</span>), which uses the No-U-Turn Sampler (NUTS) (Hoffman & Gelman, <span>2014</span>) for posterior simulation. A number of modelling design choices as well as inference approximations are made to improving mixing and computational efficiency. These are described in Appendix B.</p><p>In this section, we report some empirical evaluations of our model, which we call EpiMap. We compared two variants of EpiMap: one which models each local area separately from the rest (hence no meta-population model nor spatial component of GP), and the other the full model. For the full model we have found that the inferences are sensitive to the length scale of the spatial GP, and so we compared the full model with varying spatial length scales and with no spatial GP component. To account for uncertainty in the serial interval and incubation period distributions, we ran EpiEstim with 10 instantiations of these distributions with parameters drawn iid from the posterior distributions reported in Bi et al. (<span>2020</span>), and averaged the posterior predictive distributions over these. This procedure can be interpreted as nested Monte Carlo for a cut distribution where we specified the prior for these parameters but disallow the model from updating the prior to a posterior (Plummer, <span>2015</span>). We also compared against EpiEstim (Cori et al., <span>2013</span>) and EpiNow2 (Abbott et al., <span>2020</span>). We compared these methods on simulated data and on predicting future case counts in British local authorities. We also report estimates of <math>\n <semantics>\n <mrow>\n <msub>\n <mrow>\n <mi>R</mi>\n </mrow>\n <mrow>\n <mi>t</mi>\n </mrow>\n </msub>\n </mrow>\n <annotation>$$ {R}_t $$</annotation>\n </semantics></math> at regional and national levels.</p><p>We have proposed a hierarchical Bayesian approach to model epidemics at fine spatial scales, which incorporates movement of populations across local areas as well as spatiotemporal borrowing of strength. Empirical results suggest that our model can be a useful tool for policy-makers to locate future epidemic hotspots early, in order to direct resources such as surge testing as well as targeted local transmission reduction measures.</p><p>As with other methods that infer the extent of epidemics through identified cases alone, the main limitations of this work are due to the provenance of the Pillars 1 + 2 case data. Firstly, there can be substantial selection bias in the population who get tested, leading to discrepancies between reported cases and the true size of the epidemic. In addition, the amount of testing may change over time, for example, due to localised testing or limited supplies of testing kits, potentially leading to spurious temporal patterns (Omori et al., <span>2020</span>). Finally, case data are only reported for the combined Pillars 1 and 2 of the UK's testing regime. These correspond to different sectors of society at different points of an infection, with different delay distributions between infection and getting tested. Moreover, the proportion of tests under each pillar has been changing systematically since Pillar 2 testing began.</p><p>Our model is the result of a number of modelling choices, and can be improved in a number of ways. Firstly, our aim is to track local reproduction numbers and provide nowcasting of epidemic development in local areas, rather than understanding how NPIs affect transmission rates. This lead to our choice of a nonparametric GP prior for the reproduction numbers, rather than a generalised linear model relating transmission rates to NPIs. It is possible to extend our model to model effect of NPIs as in Flaxman et al. (<span>2020</span>). It also lead to our choice not to explicitly model the susceptible population, since it impacts the model just via lowered transmission rates.</p><p>Secondly, our model uses only Pillars 1 + 2 case data, which as noted above have biases that are not well understood. This affects our confidence in the inferred local transmission rates and forecasts. Further, in our model we assumed that positive test cases correspond 1-1 to infections, which in fact does not hold due to asymptomatic infections. We can correct for these biases by incorporating less biased data like hospitalisation and death counts, as well as less granular but better understood estimates of prevalence data obtained from randomised surveys such as REACT (Riley et al., <span>2020</span>) and the ONS infection survey (Pouwels et al., <span>2020</span>).</p><p>In order to model cross-area dependencies, we also used commuting flow data from the 2011 Census. However, this data does not necessarily reflect the commuter flow accurately during the pandemic, especially since the data is static. We used a simple approach to parameterise a time-dependent flow matrix via <math>\n <semantics>\n <mrow>\n <msub>\n <mrow>\n <mi>α</mi>\n </mrow>\n <mrow>\n <mi>t</mi>\n </mrow>\n </msub>\n </mrow>\n <annotation>$$ {\\alpha}_t $$</annotation>\n </semantics></math> which captures the overall amount of travel in each week. Nonetheless, our model is likely to improve if this limitation is addressed by using more accurate, real-time commuter flow data.</p><p>Finally, with the increasing importance of the roles of vaccines and variants, it is interesting to consider how these can be incorporated into our model. This will require a number of extensions, including separating the population into age bands and modelling the susceptible population. These extensions will incur significantly higher computational costs, and additional work will have to be performed with respect to software and implementational efficiency.</p><p>Our hierarchical Bayesian model is sensitive to a number of hyperparameters, particularly those specifying the generation interval and incubation period distributions, and the spatial and temporal length scales of the latent GP. These are hard to specify in a fully Bayesian manner. For example, the posterior strongly prefers spatial length scales that are too long due to model misspecification. Until there are good, fully Bayesian approaches to dealing with such situations, we have kept to a more pragmatic approach of using cut models and cross validation (see Section 3.1.2).</p><p>Our hierarchical model introduces stochasticity at all three layers of the model to capture different aspects of the unfolding epidemic. As a reviewer noted, there can be complex interplays between these layers, for example resulting in non-identifiable parameters. The various components of the model have been chosen to avoid the worse of these, but we have not performed a systematic study of the impacts of these choices. This will be an illuminating piece of future research.</p>","PeriodicalId":49983,"journal":{"name":"Journal of the Royal Statistical Society Series A-Statistics in Society","volume":"185 S1","pages":"S65-S85"},"PeriodicalIF":1.5000,"publicationDate":"2022-12-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://rss.onlinelibrary.wiley.com/doi/epdf/10.1111/rssa.12971","citationCount":"9","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Royal Statistical Society Series A-Statistics in Society","FirstCategoryId":"100","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/rssa.12971","RegionNum":3,"RegionCategory":"数学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"SOCIAL SCIENCES, MATHEMATICAL METHODS","Score":null,"Total":0}
引用次数: 9
Abstract
The spatio-temporal pattern of Covid-19 infections, as for most infectious disease epidemics, is highly heterogenous as a consequence of local variations in risk factors and exposures. Consequently, the widely quoted national-level estimates of reproduction numbers are of limited value in guiding local interventions and monitoring their effectiveness. It is crucial for national and local policy-makers, and for health protection teams, that accurate, well-calibrated and timely predictions of Covid-19 incidences and transmission rates are available at fine spatial scales. Obtaining such estimates is challenging, not least due to the prevalence of asymptomatic Covid-19 transmissions, as well as difficulties of obtaining high-resolution and high-frequency data. In addition, low case counts at a local level further confounds the inference for Covid-19 transmission rates, adding unwelcome uncertainty.
In this paper we develop a hierarchical Bayesian method for inference of transmission rates at fine spatial scales. Our model incorporates both temporal and spatial dependencies of local transmission rates in order to share statistical strength and reduce uncertainty. It also incorporates information about population flows to model potential transmissions across local areas. A simple approach to posterior simulation quickly becomes computationally infeasible, which is problematic if the system is required to provide timely predictions. We describe how to make posterior simulation for the model efficient, so that we are able to provide daily updates on epidemic developments.
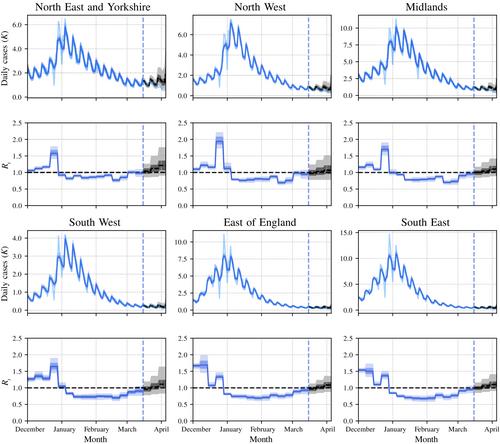
The results can be found at our web site https://localcovid.info, which is updated daily to display estimated instantaneous reproduction numbers and predicted case counts for the next weeks, across local authorities in Great Britain. The codebase updating the web site can be found at https://github.com/oxcsml/Rmap. We hope that our methodology and web site will be of interest to researchers, policy-makers and the public alike, to help identify upcoming local outbreaks and to aid in the containment of Covid-19 through both public health measures and personal decisions taken by the general public.
Our model is applied to publicly available daily counts of positive test results reported under the combined Pillars 1 (NHS and PHE) and 2 (commercial partners) of the UK's Covid-19 testing strategy.1 The data are available for 312 lower-tier local authorities (LTLAs) in England, 14 NHS Health Boards in Scotland (each covering multiple local authorities) and 22 unitary local authorities in Wales, for a total of local areas. The data are daily counts of lab-confirmed (PCR swab) cases presented by specimen date, starting from 30 January 2020. The original data are from the respective national public health authorities of England2, Scotland3and Wales4and we access them through the DELVE Global Covid-19 Dataset5 (Bhoopchand et al., 2020). Due to delays in processing tests, we ignore the last 7 days of case counts.
Our method is based on an approach to infectious disease modelling using discrete renewal processes. These have a long history, and have served as the basis for a number of recent studies estimating instantaneous reproduction numbers, (Cori et al., 2013; Flaxman et al., 2020; Fraser, 2007; Wallinga & Teunis, 2004). See Bhatt et al. (2020) and references therein for historical and mathematical background, as well as Gostic et al. (2020) for important practical considerations.
Following Flaxman et al. (2020), we model latent time series of incidence rates via renewal processes, and separate observations of reported cases using negative binomial distributions, to account for uncertainties in case reporting, outliers in case counts, and delays between infection and testing. We introduce a number of extensions and differences addressing issues that arise for applications to modelling epidemics at local authority level rather than regional or national levels. Firstly, we introduce dependencies between reproduction numbers across neighbouring localities, in order to smooth estimates of reproduction numbers and statistical strength across localities and time. We do this using a spatio-temporal Gaussian process (GP) prior for the log-transformed reproduction numbers. Secondly, we model transmissions across localities using a spatial meta-population model. Our meta-population model incorporates commuter flow data from the UK 2011 Census in order to capture stable patterns of heterogenous cross-infection rates among local authorities, linked to typical commuter patterns. Human mobility patterns may also reflect the introduction of non-pharmaceutical interventions (NPIs), though our model does not explicitly use real-time mobility data so cannot estimate the direct or indirect effects of NPIs.
The model is implemented in the Stan probabilistic programming language (Carpenter et al., 2017), which uses the No-U-Turn Sampler (NUTS) (Hoffman & Gelman, 2014) for posterior simulation. A number of modelling design choices as well as inference approximations are made to improving mixing and computational efficiency. These are described in Appendix B.
In this section, we report some empirical evaluations of our model, which we call EpiMap. We compared two variants of EpiMap: one which models each local area separately from the rest (hence no meta-population model nor spatial component of GP), and the other the full model. For the full model we have found that the inferences are sensitive to the length scale of the spatial GP, and so we compared the full model with varying spatial length scales and with no spatial GP component. To account for uncertainty in the serial interval and incubation period distributions, we ran EpiEstim with 10 instantiations of these distributions with parameters drawn iid from the posterior distributions reported in Bi et al. (2020), and averaged the posterior predictive distributions over these. This procedure can be interpreted as nested Monte Carlo for a cut distribution where we specified the prior for these parameters but disallow the model from updating the prior to a posterior (Plummer, 2015). We also compared against EpiEstim (Cori et al., 2013) and EpiNow2 (Abbott et al., 2020). We compared these methods on simulated data and on predicting future case counts in British local authorities. We also report estimates of at regional and national levels.
We have proposed a hierarchical Bayesian approach to model epidemics at fine spatial scales, which incorporates movement of populations across local areas as well as spatiotemporal borrowing of strength. Empirical results suggest that our model can be a useful tool for policy-makers to locate future epidemic hotspots early, in order to direct resources such as surge testing as well as targeted local transmission reduction measures.
As with other methods that infer the extent of epidemics through identified cases alone, the main limitations of this work are due to the provenance of the Pillars 1 + 2 case data. Firstly, there can be substantial selection bias in the population who get tested, leading to discrepancies between reported cases and the true size of the epidemic. In addition, the amount of testing may change over time, for example, due to localised testing or limited supplies of testing kits, potentially leading to spurious temporal patterns (Omori et al., 2020). Finally, case data are only reported for the combined Pillars 1 and 2 of the UK's testing regime. These correspond to different sectors of society at different points of an infection, with different delay distributions between infection and getting tested. Moreover, the proportion of tests under each pillar has been changing systematically since Pillar 2 testing began.
Our model is the result of a number of modelling choices, and can be improved in a number of ways. Firstly, our aim is to track local reproduction numbers and provide nowcasting of epidemic development in local areas, rather than understanding how NPIs affect transmission rates. This lead to our choice of a nonparametric GP prior for the reproduction numbers, rather than a generalised linear model relating transmission rates to NPIs. It is possible to extend our model to model effect of NPIs as in Flaxman et al. (2020). It also lead to our choice not to explicitly model the susceptible population, since it impacts the model just via lowered transmission rates.
Secondly, our model uses only Pillars 1 + 2 case data, which as noted above have biases that are not well understood. This affects our confidence in the inferred local transmission rates and forecasts. Further, in our model we assumed that positive test cases correspond 1-1 to infections, which in fact does not hold due to asymptomatic infections. We can correct for these biases by incorporating less biased data like hospitalisation and death counts, as well as less granular but better understood estimates of prevalence data obtained from randomised surveys such as REACT (Riley et al., 2020) and the ONS infection survey (Pouwels et al., 2020).
In order to model cross-area dependencies, we also used commuting flow data from the 2011 Census. However, this data does not necessarily reflect the commuter flow accurately during the pandemic, especially since the data is static. We used a simple approach to parameterise a time-dependent flow matrix via which captures the overall amount of travel in each week. Nonetheless, our model is likely to improve if this limitation is addressed by using more accurate, real-time commuter flow data.
Finally, with the increasing importance of the roles of vaccines and variants, it is interesting to consider how these can be incorporated into our model. This will require a number of extensions, including separating the population into age bands and modelling the susceptible population. These extensions will incur significantly higher computational costs, and additional work will have to be performed with respect to software and implementational efficiency.
Our hierarchical Bayesian model is sensitive to a number of hyperparameters, particularly those specifying the generation interval and incubation period distributions, and the spatial and temporal length scales of the latent GP. These are hard to specify in a fully Bayesian manner. For example, the posterior strongly prefers spatial length scales that are too long due to model misspecification. Until there are good, fully Bayesian approaches to dealing with such situations, we have kept to a more pragmatic approach of using cut models and cross validation (see Section 3.1.2).
Our hierarchical model introduces stochasticity at all three layers of the model to capture different aspects of the unfolding epidemic. As a reviewer noted, there can be complex interplays between these layers, for example resulting in non-identifiable parameters. The various components of the model have been chosen to avoid the worse of these, but we have not performed a systematic study of the impacts of these choices. This will be an illuminating piece of future research.
我们使用了一种简单的方法,通过α t $$ {\alpha}_t $$来参数化随时间变化的流量矩阵,该矩阵捕获了每周的总旅行量。尽管如此,如果通过使用更准确、实时的通勤流量数据来解决这一限制,我们的模型可能会得到改进。最后,随着疫苗和变异的作用日益重要,考虑如何将它们纳入我们的模型是很有趣的。这将需要许多扩展,包括将人口划分为不同的年龄组,并对易感人群进行建模。这些扩展将导致更高的计算成本,并且必须在软件和实现效率方面执行额外的工作。我们的分层贝叶斯模型对许多超参数很敏感,特别是那些指定产生间隔和潜伏期分布的超参数,以及潜在GP的空间和时间长度尺度。这些很难用完全贝叶斯的方式来定义。例如,后验强烈倾向于由于模型错误规范而导致的太长的空间长度尺度。在有好的、完全的贝叶斯方法来处理这种情况之前,我们一直使用更实用的方法,即使用切割模型和交叉验证(参见第3.1.2节)。我们的分层模型在模型的所有三层都引入了随机性,以捕捉正在展开的流行病的不同方面。正如审稿人所指出的,这些层之间可能存在复杂的相互作用,例如导致不可识别的参数。选择模型的各个组成部分是为了避免最坏的情况,但我们还没有对这些选择的影响进行系统的研究。这将是未来研究的一个有启发性的部分。
期刊介绍:
Series A (Statistics in Society) publishes high quality papers that demonstrate how statistical thinking, design and analyses play a vital role in all walks of life and benefit society in general. There is no restriction on subject-matter: any interesting, topical and revelatory applications of statistics are welcome. For example, important applications of statistical and related data science methodology in medicine, business and commerce, industry, economics and finance, education and teaching, physical and biomedical sciences, the environment, the law, government and politics, demography, psychology, sociology and sport all fall within the journal''s remit. The journal is therefore aimed at a wide statistical audience and at professional statisticians in particular. Its emphasis is on well-written and clearly reasoned quantitative approaches to problems in the real world rather than the exposition of technical detail. Thus, although the methodological basis of papers must be sound and adequately explained, methodology per se should not be the main focus of a Series A paper. Of particular interest are papers on topical or contentious statistical issues, papers which give reviews or exposés of current statistical concerns and papers which demonstrate how appropriate statistical thinking has contributed to our understanding of important substantive questions. Historical, professional and biographical contributions are also welcome, as are discussions of methods of data collection and of ethical issues, provided that all such papers have substantial statistical relevance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


