Jacqueline Kuan, Mariia Radaeva, Adeline Avenido, Artem Cherkasov, Francesco Gentile
{"title":"Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening","authors":"Jacqueline Kuan, Mariia Radaeva, Adeline Avenido, Artem Cherkasov, Francesco Gentile","doi":"10.1002/wcms.1678","DOIUrl":null,"url":null,"abstract":"<p>Recent efforts to synthetically expand drug-like chemical libraries have led to the emergence of unprecedently large virtual databases. This surge of make-on-demand molecular datasets has been received enthusiastically across the drug discovery community as a new paradigm. In several recent studies, virtual screening (VS) of larger make-on-demand collections resulted in the identification of novel molecules with higher potency and specificity compared to more conventional VS campaigns relying on smaller in-stock libraries. These results inspired ultra-large VS against various clinically relevant targets, including key proteins of the SARS-CoV-2 virus. As library sizes rapidly surpassed the billion compounds mark, new computational screening strategies emerged, shifting from conventional docking to fragment-based and machine learning-accelerated methods. These approaches significantly reduce computational demands of ultra-large screenings by lowering the number of molecules explicitly docked onto a target. Such strategies already demonstrated promise in evaluating libraries of tens of billions of molecules at relatively low computational cost. Herein, we review recent advancements in structure-based methods for ultra-large virtual screening that drug discovery practitioners have adopted to explore the ever-expanding chemical universe.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 6","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2023-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1678","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 1
Abstract
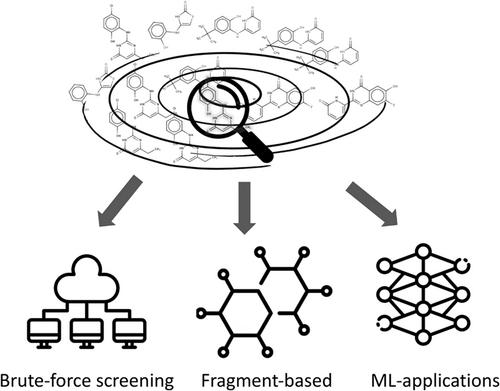
Recent efforts to synthetically expand drug-like chemical libraries have led to the emergence of unprecedently large virtual databases. This surge of make-on-demand molecular datasets has been received enthusiastically across the drug discovery community as a new paradigm. In several recent studies, virtual screening (VS) of larger make-on-demand collections resulted in the identification of novel molecules with higher potency and specificity compared to more conventional VS campaigns relying on smaller in-stock libraries. These results inspired ultra-large VS against various clinically relevant targets, including key proteins of the SARS-CoV-2 virus. As library sizes rapidly surpassed the billion compounds mark, new computational screening strategies emerged, shifting from conventional docking to fragment-based and machine learning-accelerated methods. These approaches significantly reduce computational demands of ultra-large screenings by lowering the number of molecules explicitly docked onto a target. Such strategies already demonstrated promise in evaluating libraries of tens of billions of molecules at relatively low computational cost. Herein, we review recent advancements in structure-based methods for ultra-large virtual screening that drug discovery practitioners have adopted to explore the ever-expanding chemical universe.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


