Amanda B Alvarenga, Kelli J Retallick, Andre Garcia, Stephen P Miller, Andrew Byrne, Hinayah R Oliveira, Luiz F Brito
{"title":"Across-country genetic and genomic analyses of foot score traits in American and Australian Angus cattle.","authors":"Amanda B Alvarenga, Kelli J Retallick, Andre Garcia, Stephen P Miller, Andrew Byrne, Hinayah R Oliveira, Luiz F Brito","doi":"10.1186/s12711-023-00850-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hoof structure and health are essential for the welfare and productivity of beef cattle. Therefore, we assessed the genetic and genomic background of foot score traits in American (US) and Australian (AU) Angus cattle and investigated the feasibility of performing genomic evaluations combining data for foot score traits recorded in US and AU Angus cattle. The traits evaluated were foot angle (FA) and claw set (CS). In total, 109,294 and ~ 1.12 million animals had phenotypic and genomic information, respectively. Four sets of analyses were performed: (1) genomic connectedness between US and AU Angus cattle populations and population structure, (2) estimation of genetic parameters, (3) single-step genomic prediction of breeding values, and (4) single-step genome-wide association studies for FA and CS.</p><p><strong>Results: </strong>There was no clear genetic differentiation between US and AU Angus populations. Similar heritability estimates (FA: 0.22-0.24 and CS: 0.22-0.27) and moderate-to-high genetic correlations between US and AU foot scores (FA: 0.61 and CS: 0.76) were obtained. A joint-genomic prediction using data from both populations outperformed within-country genomic evaluations. A genomic prediction model considering US and AU datasets as a single population performed similarly to the scenario accounting for genotype-by-environment interactions (i.e., multiple-trait model considering US and AU records as different traits), even though the genetic correlations between countries were lower than 0.80. Common significant genomic regions were observed between US and AU for FA and CS. Significant single nucleotide polymorphisms were identified on the Bos taurus (BTA) chromosomes BTA1, BTA5, BTA11, BTA13, BTA19, BTA20, and BTA23. The candidate genes identified were primarily from growth factor gene families, including FGF12 and GDF5, which were previously associated with bone structure and repair.</p><p><strong>Conclusions: </strong>This study presents comprehensive population structure and genetic and genomic analyses of foot scores in US and AU Angus cattle populations, which are essential for optimizing the implementation of genomic selection for improved foot scores in Angus cattle breeding programs. We have also identified candidate genes associated with foot scores in the largest Angus cattle populations in the world and made recommendations for genomic evaluations for improved foot score traits in the US and AU.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"76"},"PeriodicalIF":3.6000,"publicationDate":"2023-11-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10621155/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00850-x","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
引用次数: 0
Abstract
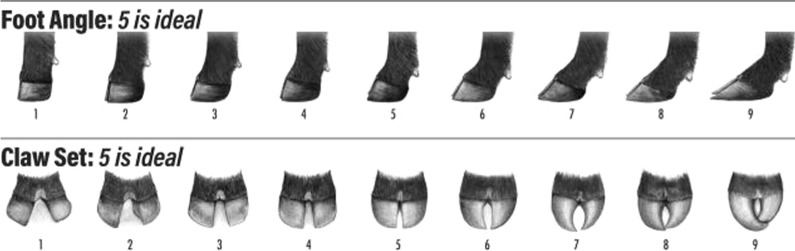
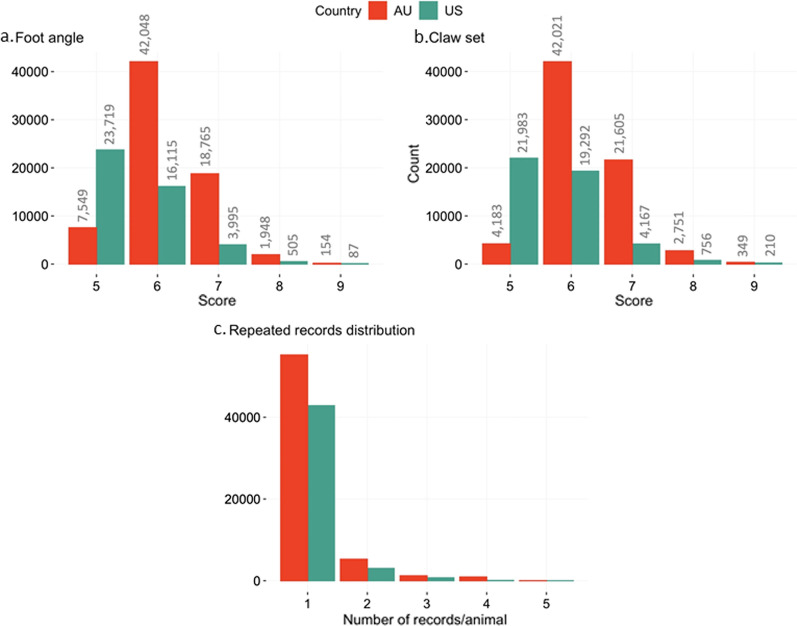
Background: Hoof structure and health are essential for the welfare and productivity of beef cattle. Therefore, we assessed the genetic and genomic background of foot score traits in American (US) and Australian (AU) Angus cattle and investigated the feasibility of performing genomic evaluations combining data for foot score traits recorded in US and AU Angus cattle. The traits evaluated were foot angle (FA) and claw set (CS). In total, 109,294 and ~ 1.12 million animals had phenotypic and genomic information, respectively. Four sets of analyses were performed: (1) genomic connectedness between US and AU Angus cattle populations and population structure, (2) estimation of genetic parameters, (3) single-step genomic prediction of breeding values, and (4) single-step genome-wide association studies for FA and CS.
Results: There was no clear genetic differentiation between US and AU Angus populations. Similar heritability estimates (FA: 0.22-0.24 and CS: 0.22-0.27) and moderate-to-high genetic correlations between US and AU foot scores (FA: 0.61 and CS: 0.76) were obtained. A joint-genomic prediction using data from both populations outperformed within-country genomic evaluations. A genomic prediction model considering US and AU datasets as a single population performed similarly to the scenario accounting for genotype-by-environment interactions (i.e., multiple-trait model considering US and AU records as different traits), even though the genetic correlations between countries were lower than 0.80. Common significant genomic regions were observed between US and AU for FA and CS. Significant single nucleotide polymorphisms were identified on the Bos taurus (BTA) chromosomes BTA1, BTA5, BTA11, BTA13, BTA19, BTA20, and BTA23. The candidate genes identified were primarily from growth factor gene families, including FGF12 and GDF5, which were previously associated with bone structure and repair.
Conclusions: This study presents comprehensive population structure and genetic and genomic analyses of foot scores in US and AU Angus cattle populations, which are essential for optimizing the implementation of genomic selection for improved foot scores in Angus cattle breeding programs. We have also identified candidate genes associated with foot scores in the largest Angus cattle populations in the world and made recommendations for genomic evaluations for improved foot score traits in the US and AU.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


