The bad and the good of trends in model building and refinement for sparse-data regions: pernicious forms of overfitting versus good new tools and predictions.
Jane S Richardson, Christopher J Williams, Vincent B Chen, Michael G Prisant, David C Richardson
{"title":"The bad and the good of trends in model building and refinement for sparse-data regions: pernicious forms of overfitting versus good new tools and predictions.","authors":"Jane S Richardson, Christopher J Williams, Vincent B Chen, Michael G Prisant, David C Richardson","doi":"10.1107/S2059798323008847","DOIUrl":null,"url":null,"abstract":"<p><p>Model building and refinement, and the validation of their correctness, are very effective and reliable at local resolutions better than about 2.5 Å for both crystallography and cryo-EM. However, at local resolutions worse than 2.5 Å both the procedures and their validation break down and do not ensure reliably correct models. This is because in the broad density at lower resolution, critical features such as protein backbone carbonyl O atoms are not just less accurate but are not seen at all, and so peptide orientations are frequently wrongly fitted by 90-180°. This puts both backbone and side chains into the wrong local energy minimum, and they are then worsened rather than improved by further refinement into a valid but incorrect rotamer or Ramachandran region. On the positive side, new tools are being developed to locate this type of pernicious error in PDB depositions, such as CaBLAM, EMRinger, Pperp diagnosis of ribose puckers, and peptide flips in PDB-REDO, while interactive modeling in Coot or ISOLDE can help to fix many of them. Another positive trend is that artificial intelligence predictions such as those made by AlphaFold2 contribute additional evidence from large multiple sequence alignments, and in high-confidence parts they provide quite good starting models for loops, termini or whole domains with otherwise ambiguous density.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":" ","pages":"1071-1078"},"PeriodicalIF":3.8000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10833350/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798323008847","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
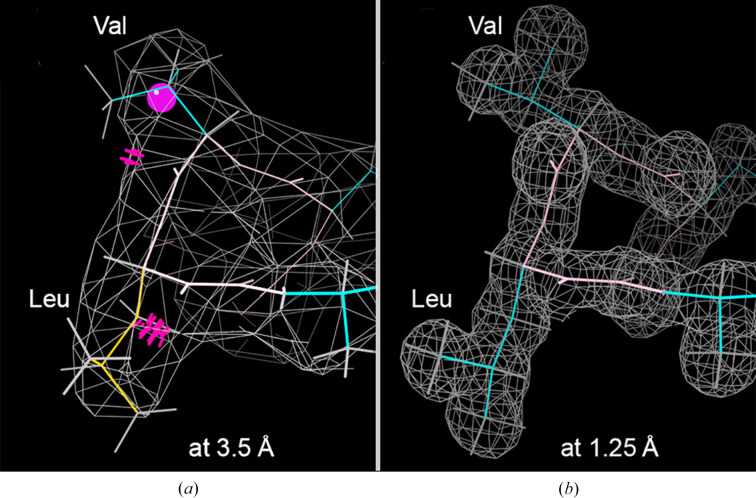
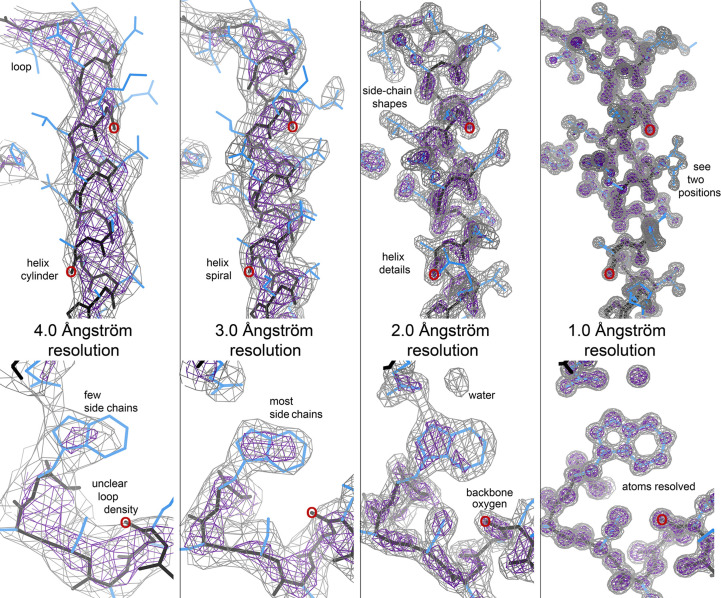
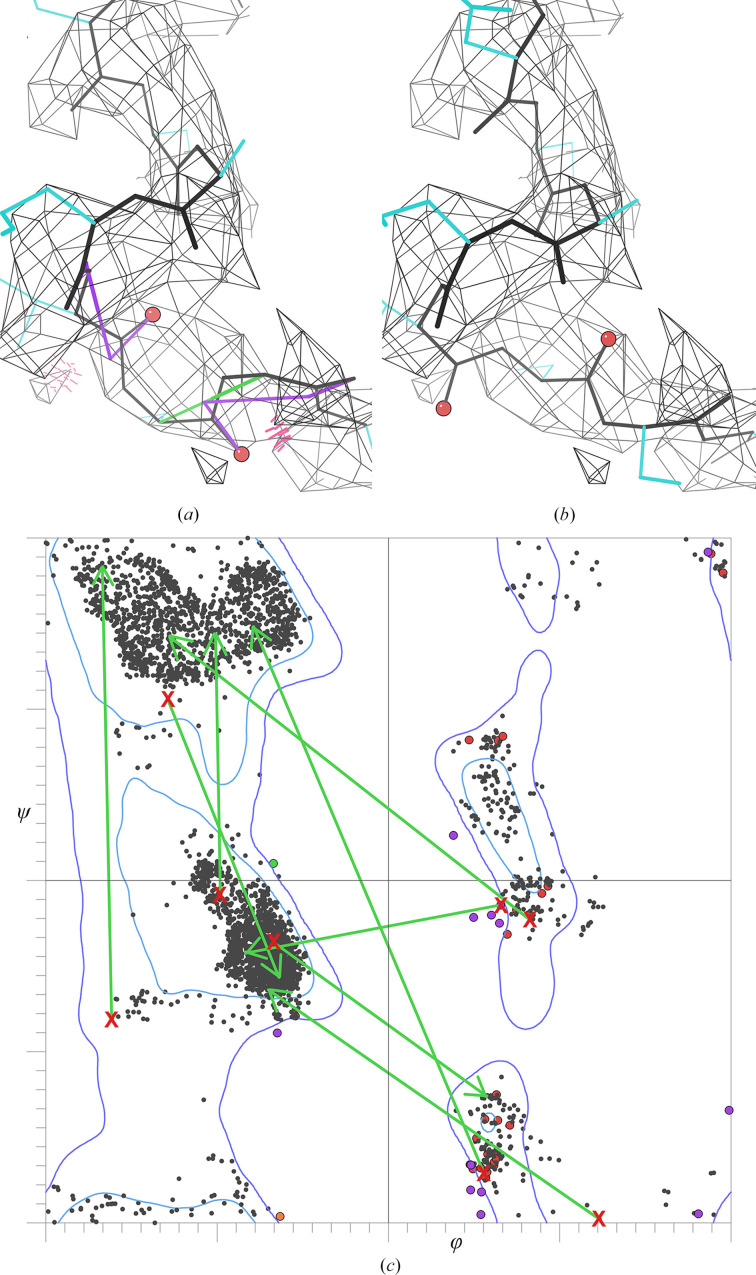
Model building and refinement, and the validation of their correctness, are very effective and reliable at local resolutions better than about 2.5 Å for both crystallography and cryo-EM. However, at local resolutions worse than 2.5 Å both the procedures and their validation break down and do not ensure reliably correct models. This is because in the broad density at lower resolution, critical features such as protein backbone carbonyl O atoms are not just less accurate but are not seen at all, and so peptide orientations are frequently wrongly fitted by 90-180°. This puts both backbone and side chains into the wrong local energy minimum, and they are then worsened rather than improved by further refinement into a valid but incorrect rotamer or Ramachandran region. On the positive side, new tools are being developed to locate this type of pernicious error in PDB depositions, such as CaBLAM, EMRinger, Pperp diagnosis of ribose puckers, and peptide flips in PDB-REDO, while interactive modeling in Coot or ISOLDE can help to fix many of them. Another positive trend is that artificial intelligence predictions such as those made by AlphaFold2 contribute additional evidence from large multiple sequence alignments, and in high-confidence parts they provide quite good starting models for loops, termini or whole domains with otherwise ambiguous density.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


