Three Pediatric Patients with Congenital Nephrogenic Diabetes Insipidus due to AVPR2 Nonsense Mutations and Different Clinical Manifestations: A Case Report.
{"title":"Three Pediatric Patients with Congenital Nephrogenic Diabetes Insipidus due to <i>AVPR2</i> Nonsense Mutations and Different Clinical Manifestations: A Case Report.","authors":"Hijiri Watanabe, Hiroshi Tamura, Keishiro Furuie, Shohei Kuraoka, Hitoshi Nakazato","doi":"10.1159/000533895","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital nephrogenic diabetes insipidus (CNDI), a rare hereditary disorder, is characterized by the inability of the kidneys to concentrate urine in response to the antidiuretic hormone arginine vasopressin (AVP); as a result, large volumes of unconcentrated urine are excreted. In addition to the clinical manifestations of CNDI, such as dehydration and electrolyte disturbances (hypernatremia and hyperchloremia), developmental delay can result without prompt treatment. In approximately 90% of cases, CNDI is an X-linked disease caused by mutations in the arginine vasopressin receptor 2 (<i>AVPR2</i>) gene. In approximately 9% of cases, CNDI is an autosomal recessive disease caused by mutations in the water channel protein aquaporin 2 (<i>AQP2</i>), and 1% of cases are autosomal dominant. We report a case of CNDI caused by a novel <i>AVPR2</i> nonsense mutation, c.520C>T (p.Q174X), and cases of siblings in another family who had a different <i>AVPR2</i> nonsense mutation, c.852G>A (p.W284X). Both cases responded well to treatment with hydrochlorothiazide and spironolactone. If CNDI is suspected, especially in carriers and neonates, aggressive genetic testing and early treatment may alleviate growth disorders and prevent irreversible central nervous system disorders and developmental delay.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"13 1","pages":"162-172"},"PeriodicalIF":0.9000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601857/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000533895","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
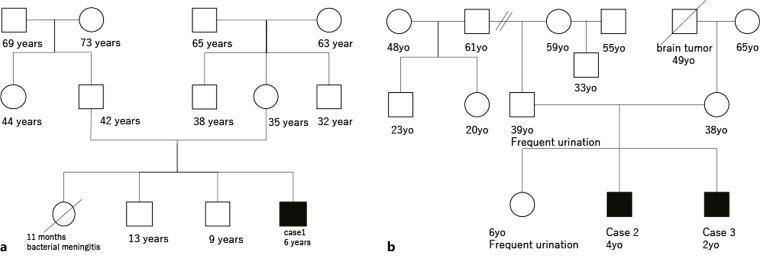
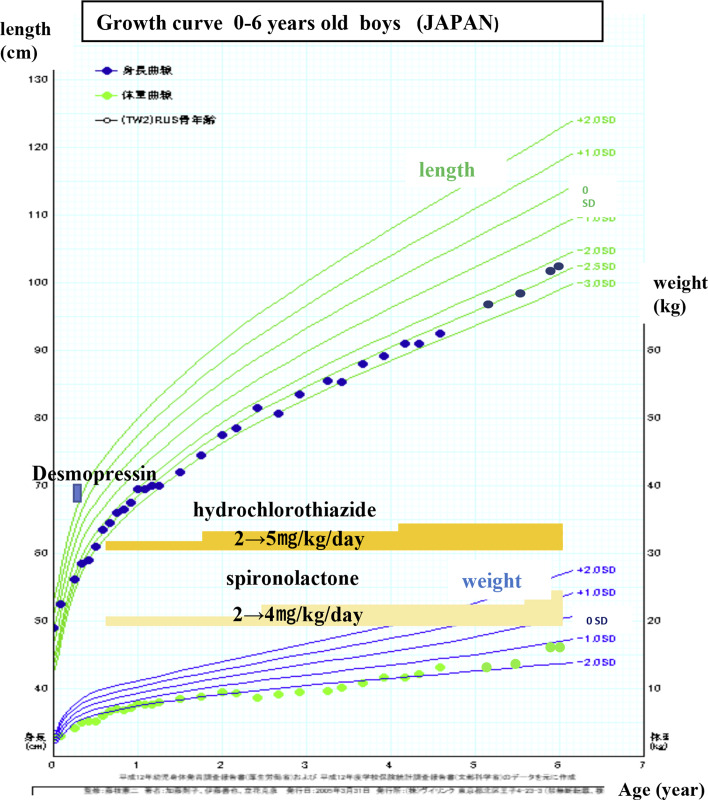
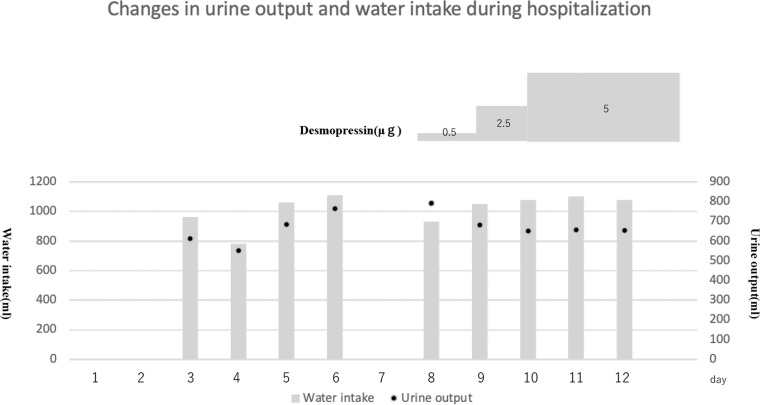
Congenital nephrogenic diabetes insipidus (CNDI), a rare hereditary disorder, is characterized by the inability of the kidneys to concentrate urine in response to the antidiuretic hormone arginine vasopressin (AVP); as a result, large volumes of unconcentrated urine are excreted. In addition to the clinical manifestations of CNDI, such as dehydration and electrolyte disturbances (hypernatremia and hyperchloremia), developmental delay can result without prompt treatment. In approximately 90% of cases, CNDI is an X-linked disease caused by mutations in the arginine vasopressin receptor 2 (AVPR2) gene. In approximately 9% of cases, CNDI is an autosomal recessive disease caused by mutations in the water channel protein aquaporin 2 (AQP2), and 1% of cases are autosomal dominant. We report a case of CNDI caused by a novel AVPR2 nonsense mutation, c.520C>T (p.Q174X), and cases of siblings in another family who had a different AVPR2 nonsense mutation, c.852G>A (p.W284X). Both cases responded well to treatment with hydrochlorothiazide and spironolactone. If CNDI is suspected, especially in carriers and neonates, aggressive genetic testing and early treatment may alleviate growth disorders and prevent irreversible central nervous system disorders and developmental delay.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


