Vadim I. Sultanov, Vadim V. Atrazhev, Dmitry V. Dmitriev
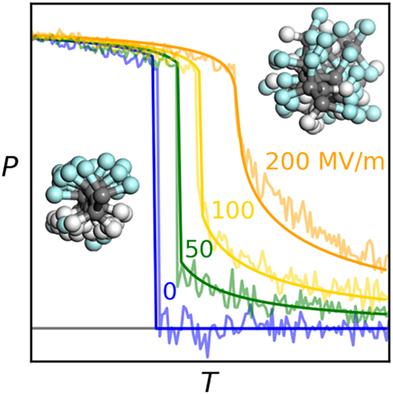
{"title":"Combined analytical and molecular dynamics model of electrocaloric effect in poly(VDF-co-TrFE) copolymer","authors":"Vadim I. Sultanov, Vadim V. Atrazhev, Dmitry V. Dmitriev","doi":"10.1002/pol.20230153","DOIUrl":null,"url":null,"abstract":"<p>A combined analytical and molecular dynamics model for the electrocaloric effect in ferroelectric poly(vinylidene difluoride-<i>co</i>-trifluoroethylene) copolymer (poly(VDF-<i>co</i>-TrFE)) is developed. The model calculates the polymer polarization, <math>\n <mrow>\n <mi>P</mi>\n </mrow></math>, and temperature change under adiabatic electric field variation, <math>\n <mrow>\n <mi>Δ</mi>\n <mi>T</mi>\n </mrow></math>, as functions of temperature. An analytical component of the model is based on the Landau phenomenological theory adapted for modeling of the first order phase transitions in a polymer crystal from a ferroelectric β phase to a paraelectric conformationally disordered (condis) phase. Parameters of the free energy functional are calibrated through molecular dynamics simulations of poly(VDF-<i>co</i>-TrFE) perfect crystal. Random orientation and the scatter of the phase transition temperature for various crystallites in a real amorphous-crystalline polymer are incorporated into the model. Comparison of the model prediction with experimental data shows good agreement for <math>\n <mrow>\n <mi>P</mi>\n <mfenced>\n <mi>T</mi>\n </mfenced>\n </mrow></math> while the model overestimates the value of <math>\n <mrow>\n <mi>Δ</mi>\n <mi>T</mi>\n </mrow></math> by approximately 2.5 times. We attribute this discrepancy to the presence of structural defects in real polymer crystallites, which reduces the entropy change under the phase transition compared to the perfect crystal simulated in our molecular dynamics approach. The theoretical limit of <math>\n <mrow>\n <mi>Δ</mi>\n <mi>T</mi>\n </mrow></math> calculated by the model indicates that <math>\n <mrow>\n <mi>Δ</mi>\n <mi>T</mi>\n </mrow></math> can be increased up to 3 times compared to the currently observed experimental value of <math>\n <mrow>\n <mi>Δ</mi>\n <mi>T</mi>\n </mrow></math>.</p>","PeriodicalId":199,"journal":{"name":"Journal of Polymer Science Part A: Polymer Chemistry","volume":"61 18","pages":"2091-2102"},"PeriodicalIF":2.7020,"publicationDate":"2023-06-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Polymer Science Part A: Polymer Chemistry","FirstCategoryId":"1","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/pol.20230153","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Materials Science","Score":null,"Total":0}
引用次数: 1
Abstract
A combined analytical and molecular dynamics model for the electrocaloric effect in ferroelectric poly(vinylidene difluoride-co-trifluoroethylene) copolymer (poly(VDF-co-TrFE)) is developed. The model calculates the polymer polarization, , and temperature change under adiabatic electric field variation, , as functions of temperature. An analytical component of the model is based on the Landau phenomenological theory adapted for modeling of the first order phase transitions in a polymer crystal from a ferroelectric β phase to a paraelectric conformationally disordered (condis) phase. Parameters of the free energy functional are calibrated through molecular dynamics simulations of poly(VDF-co-TrFE) perfect crystal. Random orientation and the scatter of the phase transition temperature for various crystallites in a real amorphous-crystalline polymer are incorporated into the model. Comparison of the model prediction with experimental data shows good agreement for while the model overestimates the value of by approximately 2.5 times. We attribute this discrepancy to the presence of structural defects in real polymer crystallites, which reduces the entropy change under the phase transition compared to the perfect crystal simulated in our molecular dynamics approach. The theoretical limit of calculated by the model indicates that can be increased up to 3 times compared to the currently observed experimental value of .
期刊介绍:
Part A: Polymer Chemistry is devoted to studies in fundamental organic polymer chemistry and physical organic chemistry. This includes all related topics (such as organic, bioorganic, bioinorganic and biological chemistry of monomers, polymers, oligomers and model compounds, inorganic and organometallic chemistry for catalysts, mechanistic studies, supramolecular chemistry aspects relevant to polymer...

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


