{"title":"Along the allostery stream: Recent advances in computational methods for allosteric drug discovery","authors":"Duan Ni, Zongtao Chai, Ying Wang, Mingyu Li, Zhengtian Yu, Yaqin Liu, Shaoyong Lu, Jian Zhang","doi":"10.1002/wcms.1585","DOIUrl":null,"url":null,"abstract":"<p>Allostery is a universal, biological phenomenon in which orthosteric sites are fine-tuned by topologically distal allosteric sites triggered by perturbations, such as ligand binding, residue mutations, or post-translational modifications. Allosteric regulation is implicated in a variety of physiological and pathological conditions and is thus emerging as a novel avenue for drug discovery. Allosteric drugs have traditionally been discovered by serendipity through large-scale experimental screening. Recently, we have witnessed significant progress in biophysics, particularly in structural bioinformatics, which has facilitated the in-depth characterization of allosteric effects and the accurate detection of allosteric residues and exosites. These advances improve our understanding of allosterism and promote allosteric drug discovery, thereby revolutionizing the shift from the traditional serendipitous route used to discover allosteric drugs to the updated path centered on rational structure-based design. In this review, recent advances in computational methods applied to allosteric drug discovery are summarized. We comprehensively review these achievements along various levels of allosteric events, from the construction of allosteric databases to the identification and analysis of allosteric residues, signals, sites, and modulators. We expect to increase the awareness of the discovery of allosteric drugs using structure-based computational methods.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"12 4","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2021-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"22","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1585","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 22
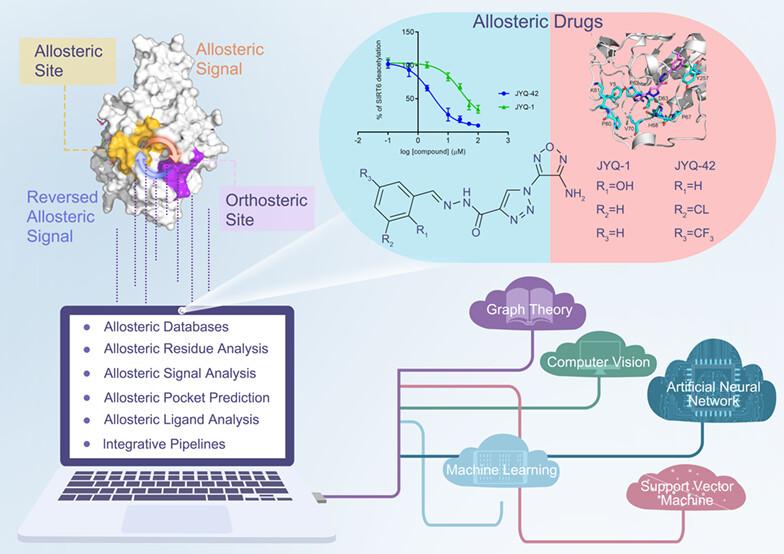
Abstract
Allostery is a universal, biological phenomenon in which orthosteric sites are fine-tuned by topologically distal allosteric sites triggered by perturbations, such as ligand binding, residue mutations, or post-translational modifications. Allosteric regulation is implicated in a variety of physiological and pathological conditions and is thus emerging as a novel avenue for drug discovery. Allosteric drugs have traditionally been discovered by serendipity through large-scale experimental screening. Recently, we have witnessed significant progress in biophysics, particularly in structural bioinformatics, which has facilitated the in-depth characterization of allosteric effects and the accurate detection of allosteric residues and exosites. These advances improve our understanding of allosterism and promote allosteric drug discovery, thereby revolutionizing the shift from the traditional serendipitous route used to discover allosteric drugs to the updated path centered on rational structure-based design. In this review, recent advances in computational methods applied to allosteric drug discovery are summarized. We comprehensively review these achievements along various levels of allosteric events, from the construction of allosteric databases to the identification and analysis of allosteric residues, signals, sites, and modulators. We expect to increase the awareness of the discovery of allosteric drugs using structure-based computational methods.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


