Workflow for large-scale analysis of melanoma tissue samples
Q4 Biochemistry, Genetics and Molecular Biology
引用次数: 6
Abstract
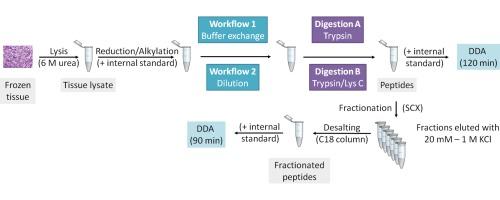
The aim of the present study was to create an optimal workflow for analysing a large cohort of malignant melanoma tissue samples. Samples were lysed with urea and enzymatically digested with trypsin or trypsin/Lys C. Buffer exchange or dilution was used to reduce urea concentration prior to digestion. The tissue digests were analysed directly or following strong cation exchange (SCX) fractionation by nano LC–MS/MS. The approach which resulted in the largest number of protein IDs involved a buffer exchange step before enzymatic digestion with trypsin and chromatographic separation in 120 min gradient followed by SCX–RP separation of peptides.
大规模分析黑色素瘤组织样本的工作流程
本研究的目的是为分析大量恶性黑色素瘤组织样本创建一个最佳工作流程。样品用尿素裂解,用胰蛋白酶或胰蛋白酶/赖氨酸c酶解。酶解前用缓冲液交换或稀释降低尿素浓度。采用纳米LC-MS /MS对组织消化液进行直接或强阳离子交换(SCX)分馏分析。在胰蛋白酶酶切之前进行缓冲交换步骤,然后以120分钟的梯度进行色谱分离,然后进行SCX-RP分离,从而获得最多的蛋白质id。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

EuPA Open Proteomics
Biochemistry, Genetics and Molecular Biology-Biochemistry
自引率
0.00%
发文量
0
审稿时长
103 days
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


