{"title":"CRISPR/Cas-based gene editing in therapeutic strategies for beta-thalassemia.","authors":"Shujun Zeng, Shuangyin Lei, Chao Qu, Yue Wang, Shuzhi Teng, Ping Huang","doi":"10.1007/s00439-023-02610-9","DOIUrl":null,"url":null,"abstract":"<p><p>Beta-thalassemia (β-thalassemia) is an autosomal recessive disorder caused by point mutations, insertions, and deletions in the HBB gene cluster, resulting in the underproduction of β-globin chains. The most severe type may demonstrate complications including massive hepatosplenomegaly, bone deformities, and severe growth retardation in children. Treatments for β-thalassemia include blood transfusion, splenectomy, and allogeneic hematopoietic stem cell transplantation (HSCT). However, long-term blood transfusions require regular iron removal therapy. For allogeneic HSCT, human lymphocyte antigen (HLA)-matched donors are rarely available, and acute graft-versus-host disease (GVHD) may occur after the transplantation. Thus, these conventional treatments are facing significant challenges. In recent years, with the advent and advancement of CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) gene editing technology, precise genome editing has achieved encouraging successes in basic and clinical studies for treating various genetic disorders, including β-thalassemia. Target gene-edited autogeneic HSCT helps patients avoid graft rejection and GVHD, making it a promising curative therapy for transfusion-dependent β-thalassemia (TDT). In this review, we introduce the development and mechanisms of CRISPR/Cas9. Recent advances on feasible strategies of CRISPR/Cas9 targeting three globin genes (HBB, HBG, and HBA) and targeting cell selections for β-thalassemia therapy are highlighted. Current CRISPR-based clinical trials in the treatment of β-thalassemia are summarized, which are focused on γ-globin reactivation and fetal hemoglobin reproduction in hematopoietic stem cells. Lastly, the applications of other promising CRISPR-based technologies, such as base editing and prime editing, in treating β-thalassemia and the limitations of the CRISPR/Cas system in therapeutic applications are discussed.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"1677-1703"},"PeriodicalIF":3.8000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-023-02610-9","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
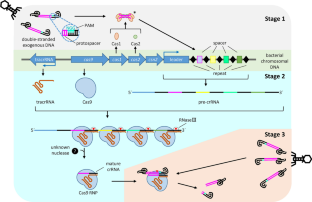
Abstract
Beta-thalassemia (β-thalassemia) is an autosomal recessive disorder caused by point mutations, insertions, and deletions in the HBB gene cluster, resulting in the underproduction of β-globin chains. The most severe type may demonstrate complications including massive hepatosplenomegaly, bone deformities, and severe growth retardation in children. Treatments for β-thalassemia include blood transfusion, splenectomy, and allogeneic hematopoietic stem cell transplantation (HSCT). However, long-term blood transfusions require regular iron removal therapy. For allogeneic HSCT, human lymphocyte antigen (HLA)-matched donors are rarely available, and acute graft-versus-host disease (GVHD) may occur after the transplantation. Thus, these conventional treatments are facing significant challenges. In recent years, with the advent and advancement of CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) gene editing technology, precise genome editing has achieved encouraging successes in basic and clinical studies for treating various genetic disorders, including β-thalassemia. Target gene-edited autogeneic HSCT helps patients avoid graft rejection and GVHD, making it a promising curative therapy for transfusion-dependent β-thalassemia (TDT). In this review, we introduce the development and mechanisms of CRISPR/Cas9. Recent advances on feasible strategies of CRISPR/Cas9 targeting three globin genes (HBB, HBG, and HBA) and targeting cell selections for β-thalassemia therapy are highlighted. Current CRISPR-based clinical trials in the treatment of β-thalassemia are summarized, which are focused on γ-globin reactivation and fetal hemoglobin reproduction in hematopoietic stem cells. Lastly, the applications of other promising CRISPR-based technologies, such as base editing and prime editing, in treating β-thalassemia and the limitations of the CRISPR/Cas system in therapeutic applications are discussed.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


