Substitution effect on the adiabatic ionization potential, vertical ionization potential, electrophilicity, and nucleophilicity of some hydantoin drug derivatives: Computational study
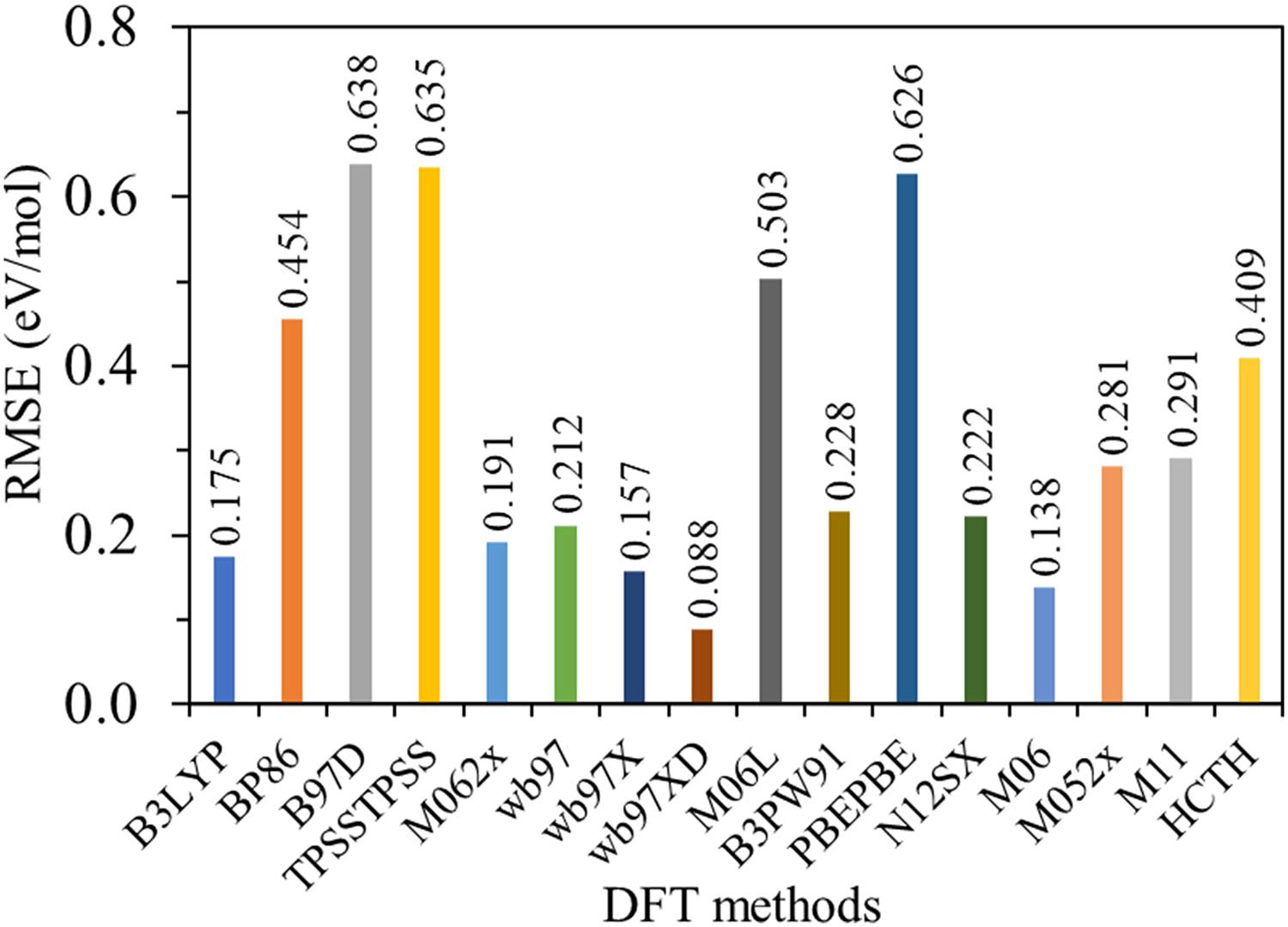
{"title":"Substitution effect on the adiabatic ionization potential, vertical ionization potential, electrophilicity, and nucleophilicity of some hydantoin drug derivatives: Computational study","authors":"Zaki Safi, Nuha Wazzan","doi":"10.1002/poc.4565","DOIUrl":null,"url":null,"abstract":"<p>In the current paper, the adiabatic ionization potentials (AIP) for 29 hydantoin derivatives and hydantoin-based drugs such as allantoin, phenytoin, mephenytoin, nilutamide, iprodione, nitrofurantoin, and ethotoin were calculated using the double hybrid ωB97XD density functional theory (DFT) in coupling with 6-311+G(2df,2p) basis set at the B3LYP/6-31+G(d,p) optimized geometry. The neutral and cationic radicals of the examined species were firstly optimized using the B3LYP/6-31+G(d,p) level. Final energies were improved by single point calculation using 16 different DFT methods such as B3LYP, ωB97, B97D, TPSSTPSS, M06-2X, …, and so forth, with 6-311+G(2df,2p) basis. Statistical tools such as root mean square error (RMSE) was used to examine the accuracy of the DFT method with respect to the standard reference AIP values. These standard references were calculated, for 12 hydantoin derivatives with less than nine non-hydrogen atoms, by taking the average values of the AIP computed using the G4, G3B3, and CBS-QBS methods. The vertical ionization potentials (VIPs), the vertical electron affinity (VEA), and global quantum parameters such as electrophilicity and nucleophilicity of the 29 molecules were also calculated. Substitution effect on the AIP, VIP, VEA, fundamental gap, electrophilicity, and nucleophilicity of the species under probe was studied and discussed. The results reveal that substitution of electron withdrawing group (EWG) raises the AIP and VIP, electrophilicity, and the fundamental gap, while substitution of electron donating group (EDG) raises the VEA and the nucleophilicity. Furthermore, the condensed Fukui functions were used to identify the active centers for nucleophilic, electrophilic, and free radical attacks.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"36 11","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2023-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4565","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
Abstract
In the current paper, the adiabatic ionization potentials (AIP) for 29 hydantoin derivatives and hydantoin-based drugs such as allantoin, phenytoin, mephenytoin, nilutamide, iprodione, nitrofurantoin, and ethotoin were calculated using the double hybrid ωB97XD density functional theory (DFT) in coupling with 6-311+G(2df,2p) basis set at the B3LYP/6-31+G(d,p) optimized geometry. The neutral and cationic radicals of the examined species were firstly optimized using the B3LYP/6-31+G(d,p) level. Final energies were improved by single point calculation using 16 different DFT methods such as B3LYP, ωB97, B97D, TPSSTPSS, M06-2X, …, and so forth, with 6-311+G(2df,2p) basis. Statistical tools such as root mean square error (RMSE) was used to examine the accuracy of the DFT method with respect to the standard reference AIP values. These standard references were calculated, for 12 hydantoin derivatives with less than nine non-hydrogen atoms, by taking the average values of the AIP computed using the G4, G3B3, and CBS-QBS methods. The vertical ionization potentials (VIPs), the vertical electron affinity (VEA), and global quantum parameters such as electrophilicity and nucleophilicity of the 29 molecules were also calculated. Substitution effect on the AIP, VIP, VEA, fundamental gap, electrophilicity, and nucleophilicity of the species under probe was studied and discussed. The results reveal that substitution of electron withdrawing group (EWG) raises the AIP and VIP, electrophilicity, and the fundamental gap, while substitution of electron donating group (EDG) raises the VEA and the nucleophilicity. Furthermore, the condensed Fukui functions were used to identify the active centers for nucleophilic, electrophilic, and free radical attacks.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


