Identification of diagnostic markers of pancreatic ductal adenocarcinoma using transcriptomic tumour and blood sample data
Abstract
Background
Pancreatic ductal adenocarcinoma (PDAC) is the most frequently diagnosed form of pancreatic cancer worldwide. PDAC is associated with a poor survival rate mainly due to the disease being usually diagnosed at late stages.
Methods
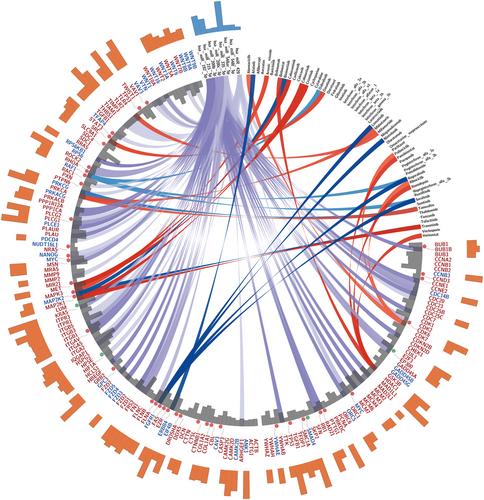
Publicly available gene expression data from 10 studies with tumour tissue (448 samples) and/or blood samples (128 samples) from PDAC patients were pooled together and analyzed for the identification of stage-specific and global diagnostic markers using differential gene expression analysis. The list of statistically significant () differentially expressed genes were used to carry out enrichment analysis via active subnetworks and miRNA enrichment analysis. We then used the results from these analyses to identify the most significant genes and pathways and map these to marketed drugs’ pharmacological targets. The same process was replicated for studies with blood samples and results were compared to those from the tissue analysis. A set of consistently deregulated genes (pancreatic tumour signature, PTS) in both tissue and blood samples was derived and validated in external cohorts and The Cancer Genome Atlas (TCGA) data.
Results
Notable gene expression deregulation was found in all tumour stages with significant overlap. We identified 820 consistently deregulated genes (PTS) in tissue samples of all stages and blood samples. Active subnetwork analysis revealed enriched ribosome, proteasome, adherens junction and cell cycle pathways across all stages and blood samples. Our findings suggest that microRNA (miRNA) contribution to PDAC pathology plays a significant role and is probably mediated by distinct miRNAs across stages of PDAC. Stage-specific enriched miRNAs with diagnostic potential included miR-21, miR-29, miR-124 and miR-30, for stages 1–4, respectively. By investigating the pharmacogenetic interactions of the identified targets with clinically approved drugs, we outline potential paths for personalized interventions. Importantly, the PTS showed a significant association with survival in TCGA data.
Conclusion
Thus, we present a compilation of protein-coding markers and miRNAs that hold potential as a diagnostic tool for the early detection of PDAC, as well as for designing novel therapeutic strategies aimed at improving patient outcomes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


