Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow.
Liang Jin, Fei Wang, Xue Wang, Bohdan P Harvey, Yingtao Bi, Chenqi Hu, Baoliang Cui, Anhdao T Darcy, John W Maull, Ben R Phillips, Youngjae Kim, Gary J Jenkins, Thierry R Sornasse, Yu Tian
{"title":"Identification of Plasma Biomarkers from Rheumatoid Arthritis Patients Using an Optimized Sequential Window Acquisition of All THeoretical Mass Spectra (SWATH) Proteomics Workflow.","authors":"Liang Jin, Fei Wang, Xue Wang, Bohdan P Harvey, Yingtao Bi, Chenqi Hu, Baoliang Cui, Anhdao T Darcy, John W Maull, Ben R Phillips, Youngjae Kim, Gary J Jenkins, Thierry R Sornasse, Yu Tian","doi":"10.3390/proteomes11040032","DOIUrl":null,"url":null,"abstract":"<p><p>Rheumatoid arthritis (RA) is a systemic autoimmune and inflammatory disease. Plasma biomarkers are critical for understanding disease mechanisms, treatment effects, and diagnosis. Mass spectrometry-based proteomics is a powerful tool for unbiased biomarker discovery. However, plasma proteomics is significantly hampered by signal interference from high-abundance proteins, low overall protein coverage, and high levels of missing data from data-dependent acquisition (DDA). To achieve quantitative proteomics analysis for plasma samples with a balance of throughput, performance, and cost, we developed a workflow incorporating plate-based high abundance protein depletion and sample preparation, comprehensive peptide spectral library building, and data-independent acquisition (DIA) SWATH mass spectrometry-based methodology. In this study, we analyzed plasma samples from both RA patients and healthy donors. The results showed that the new workflow performance exceeded that of the current state-of-the-art depletion-based plasma proteomic platforms in terms of both data quality and proteome coverage. Proteins from biological processes related to the activation of systemic inflammation, suppression of platelet function, and loss of muscle mass were enriched and differentially expressed in RA. Some plasma proteins, particularly acute-phase reactant proteins, showed great power to distinguish between RA patients and healthy donors. Moreover, protein isoforms in the plasma were also analyzed, providing even deeper proteome coverage. This workflow can serve as a basis for further application in discovering plasma biomarkers of other diseases.</p>","PeriodicalId":20877,"journal":{"name":"Proteomes","volume":"11 4","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2023-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10594463/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/proteomes11040032","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
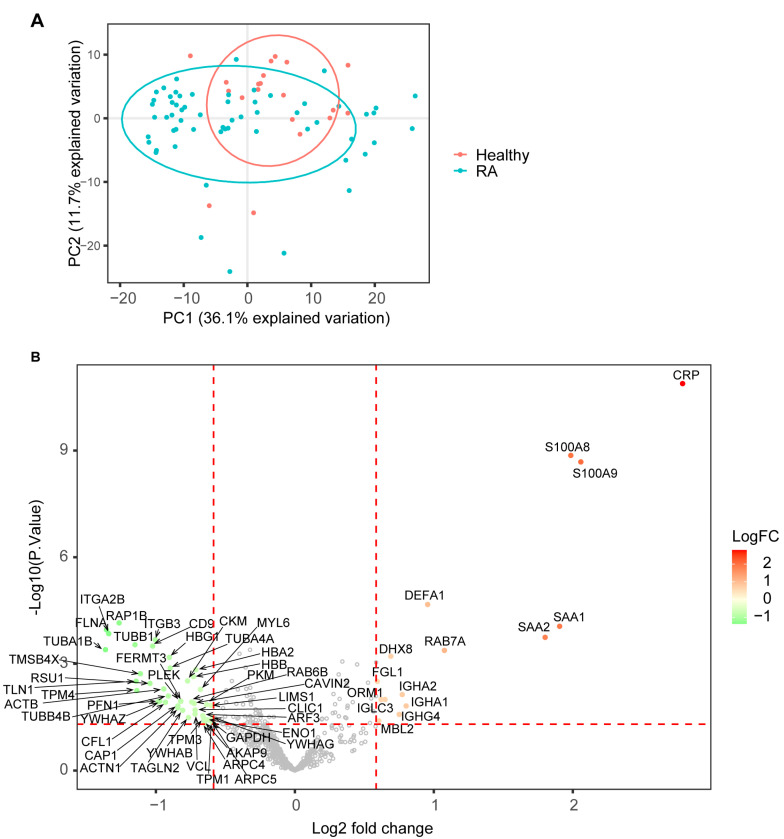
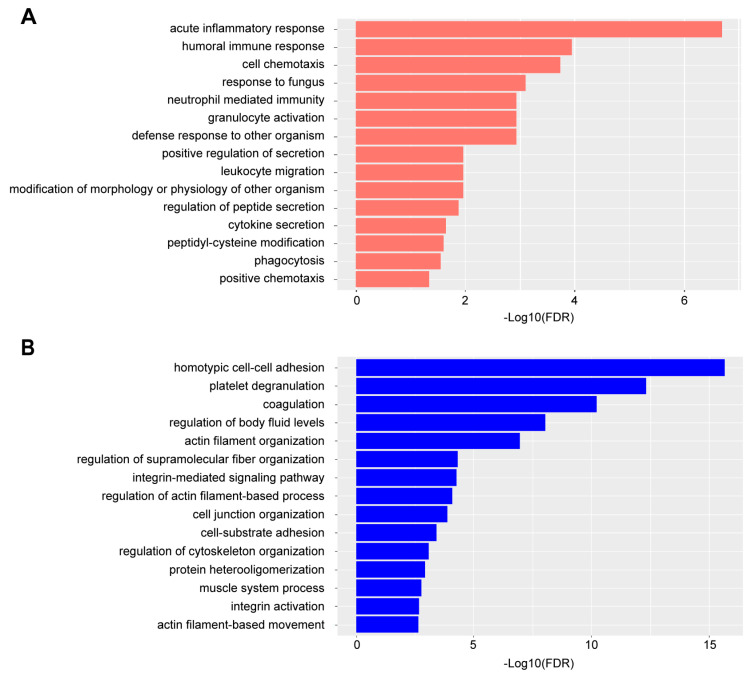
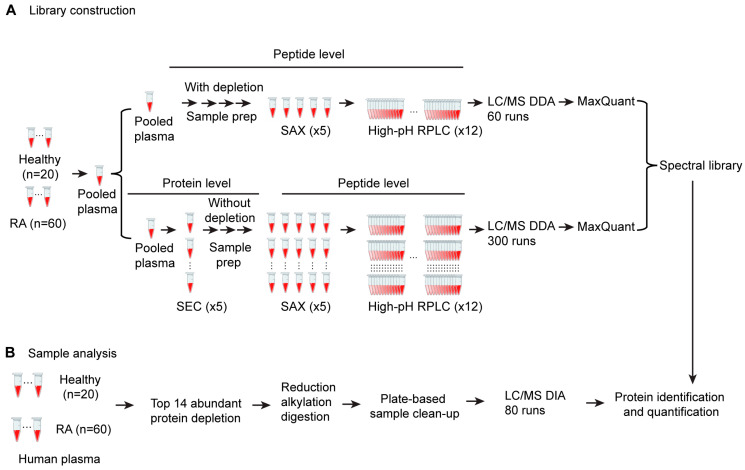
Rheumatoid arthritis (RA) is a systemic autoimmune and inflammatory disease. Plasma biomarkers are critical for understanding disease mechanisms, treatment effects, and diagnosis. Mass spectrometry-based proteomics is a powerful tool for unbiased biomarker discovery. However, plasma proteomics is significantly hampered by signal interference from high-abundance proteins, low overall protein coverage, and high levels of missing data from data-dependent acquisition (DDA). To achieve quantitative proteomics analysis for plasma samples with a balance of throughput, performance, and cost, we developed a workflow incorporating plate-based high abundance protein depletion and sample preparation, comprehensive peptide spectral library building, and data-independent acquisition (DIA) SWATH mass spectrometry-based methodology. In this study, we analyzed plasma samples from both RA patients and healthy donors. The results showed that the new workflow performance exceeded that of the current state-of-the-art depletion-based plasma proteomic platforms in terms of both data quality and proteome coverage. Proteins from biological processes related to the activation of systemic inflammation, suppression of platelet function, and loss of muscle mass were enriched and differentially expressed in RA. Some plasma proteins, particularly acute-phase reactant proteins, showed great power to distinguish between RA patients and healthy donors. Moreover, protein isoforms in the plasma were also analyzed, providing even deeper proteome coverage. This workflow can serve as a basis for further application in discovering plasma biomarkers of other diseases.
ProteomesBiochemistry, Genetics and Molecular Biology-Clinical Biochemistry
CiteScore
6.50
自引率
3.00%
发文量
37
审稿时长
11 weeks
期刊介绍:
Proteomes (ISSN 2227-7382) is an open access, peer reviewed journal on all aspects of proteome science. Proteomes covers the multi-disciplinary topics of structural and functional biology, protein chemistry, cell biology, methodology used for protein analysis, including mass spectrometry, protein arrays, bioinformatics, HTS assays, etc. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. Therefore, there is no restriction on the length of papers. Scope: -whole proteome analysis of any organism -disease/pharmaceutical studies -comparative proteomics -protein-ligand/protein interactions -structure/functional proteomics -gene expression -methodology -bioinformatics -applications of proteomics

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


