{"title":"A new interatomic potential describing Fe-H and H-H interactions in bcc iron","authors":"Mao Wen","doi":"10.1016/j.commatsci.2021.110640","DOIUrl":null,"url":null,"abstract":"<div><p>We present a new many-body interatomic potential<span> for H in body-centered cubic (bcc) Fe. The potential is developed based on extensive energetics and atomic configurations of an H atom and H-H interactions in Fe from density functional theory calculations. In detail, the potential is parameterized by fitting not only to a single H atom in the perfect bcc Fe lattice and to the properties of H trap binding to a vacancy and surfaces as being done by previous studies, but also to multiple H trapping to a vacancy and H-H interaction in Fe lattice. With such a fitting strategy, the developed potential outperforms existing potentials in its ability not only describing the behaviors of a single H atom in Fe, but also capturing the features of H-H interaction reliably, which is of key importance in revealing H behaviors in local H accumulation around dislocation cores, grain boundaries and crack tips.</span></p></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"197 ","pages":"Article 110640"},"PeriodicalIF":3.3000,"publicationDate":"2021-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.commatsci.2021.110640","citationCount":"12","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025621003670","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 12
Abstract
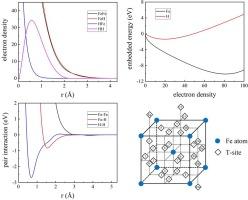
We present a new many-body interatomic potential for H in body-centered cubic (bcc) Fe. The potential is developed based on extensive energetics and atomic configurations of an H atom and H-H interactions in Fe from density functional theory calculations. In detail, the potential is parameterized by fitting not only to a single H atom in the perfect bcc Fe lattice and to the properties of H trap binding to a vacancy and surfaces as being done by previous studies, but also to multiple H trapping to a vacancy and H-H interaction in Fe lattice. With such a fitting strategy, the developed potential outperforms existing potentials in its ability not only describing the behaviors of a single H atom in Fe, but also capturing the features of H-H interaction reliably, which is of key importance in revealing H behaviors in local H accumulation around dislocation cores, grain boundaries and crack tips.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


