{"title":"New interatomic potential for simulation of pure magnesium and magnesium hydrides","authors":"D.E. Smirnova , S.V. Starikov , A.M. Vlasova","doi":"10.1016/j.commatsci.2018.07.051","DOIUrl":null,"url":null,"abstract":"<div><p>We develop an interatomic potential intended for the study of Mg-H system using atomistic methods. The reported potential has an angular-dependent form and can be used for simulation of pure magnesium, as well as for consideration of binary cases including Mg and H. Summary of the performed tests on elastic, thermophysical and diffusional properties proves that the potential has a wide range of applicability. For example, it can be used to model phase transitions existing in pure magnesium (liquid <span><math><mrow><mo>↔</mo></mrow></math></span> hcp and bcc <span><math><mrow><mo>↔</mo></mrow></math></span> hcp). We also show how the model represents energies of different point defects and stacking faults in Mg. The primary purpose of the potential is the simulation of hydrogen behavior in magnesium. Here we show examples of the hydrogen diffusion and clusterization in hcp magnesium. Also, it is shown that the proposed potential reproduces stable structures for some of the existing magnesium hydrides: <span><math><mrow><mi>α</mi></mrow></math></span>-MgH<sub>2</sub> (P4<sub>2</sub>/mnm) and <span><math><mrow><mi>γ</mi></mrow></math></span>-MgH<sub>2</sub> (Pbcn).</p></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"154 ","pages":"Pages 295-302"},"PeriodicalIF":3.1000,"publicationDate":"2018-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.commatsci.2018.07.051","citationCount":"17","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025618304865","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 17
Abstract
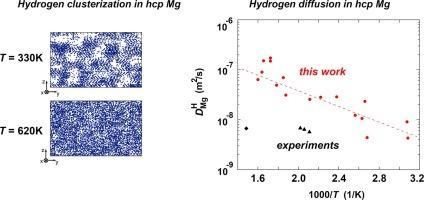
We develop an interatomic potential intended for the study of Mg-H system using atomistic methods. The reported potential has an angular-dependent form and can be used for simulation of pure magnesium, as well as for consideration of binary cases including Mg and H. Summary of the performed tests on elastic, thermophysical and diffusional properties proves that the potential has a wide range of applicability. For example, it can be used to model phase transitions existing in pure magnesium (liquid hcp and bcc hcp). We also show how the model represents energies of different point defects and stacking faults in Mg. The primary purpose of the potential is the simulation of hydrogen behavior in magnesium. Here we show examples of the hydrogen diffusion and clusterization in hcp magnesium. Also, it is shown that the proposed potential reproduces stable structures for some of the existing magnesium hydrides: -MgH2 (P42/mnm) and -MgH2 (Pbcn).
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


