Yingchao Su, Zhihui Li, Xinming Rang, Yifei Wang, Jin Fu
{"title":"Integrated Analysis and Identification of CSF-Derived Risk miRNAs and Pivotal Genes in Multiple Sclerosis","authors":"Yingchao Su, Zhihui Li, Xinming Rang, Yifei Wang, Jin Fu","doi":"10.1007/s12031-022-02007-9","DOIUrl":null,"url":null,"abstract":"<div><p>Multiple sclerosis (MS) is a common chronic autoimmune disorder of the central nervous system that predominantly affects young adults. Mounting evidence indicates that deregulation of microRNAs (miRNAs) in cerebrospinal fluid (CSF) has been implicated in MS as a potential biomarker. However, comprehensive assessments of CSF miRNAs and their target genes are lacking. Here, aberrantly expressed CSF miRNAs of MS patients were obtained from numerous studies by manual search. With detailed information on these miRNAs, we utilized online databases to screen out immune-related target genes and further performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. To identify MS high-risk pathways and pivotal genes, pathway crosstalk and pathway-gene networks were constructed, followed by the establishment of a protein–protein interaction (PPI) network. The datasets collected from ArrayExpress were used to assess pivotal genes. Overall, 21 MS-related CSF miRNAs were included in this study. Subsequently, we identified 469 MS-related genes and 14 high-risk pathways. In the pathway-gene network, 27 critical MS-related genes participated in at least half of the high-risk pathways, and these genes were used to identify pivotal genes. Finally, miR-150, miR-328, and miR-34c-5p were determined to be risk miRNAs via the regulation of the pivotal risk genes MAPK1, AKT1, and VEGFA. Among them, VEGFA was validated to be significantly decreased in the CSF cells of MS patients by transcriptomic datasets. These findings may provide potential biomarkers or therapeutic targets and help elucidate the molecular mechanisms underlying the pathogenesis of MS.</p></div>","PeriodicalId":652,"journal":{"name":"Journal of Molecular Neuroscience","volume":"72 9","pages":"1916 - 1928"},"PeriodicalIF":2.8000,"publicationDate":"2022-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Neuroscience","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s12031-022-02007-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
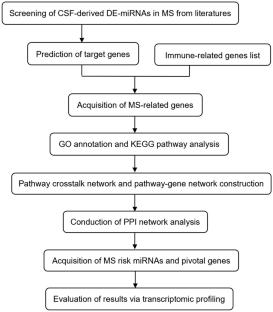
Multiple sclerosis (MS) is a common chronic autoimmune disorder of the central nervous system that predominantly affects young adults. Mounting evidence indicates that deregulation of microRNAs (miRNAs) in cerebrospinal fluid (CSF) has been implicated in MS as a potential biomarker. However, comprehensive assessments of CSF miRNAs and their target genes are lacking. Here, aberrantly expressed CSF miRNAs of MS patients were obtained from numerous studies by manual search. With detailed information on these miRNAs, we utilized online databases to screen out immune-related target genes and further performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. To identify MS high-risk pathways and pivotal genes, pathway crosstalk and pathway-gene networks were constructed, followed by the establishment of a protein–protein interaction (PPI) network. The datasets collected from ArrayExpress were used to assess pivotal genes. Overall, 21 MS-related CSF miRNAs were included in this study. Subsequently, we identified 469 MS-related genes and 14 high-risk pathways. In the pathway-gene network, 27 critical MS-related genes participated in at least half of the high-risk pathways, and these genes were used to identify pivotal genes. Finally, miR-150, miR-328, and miR-34c-5p were determined to be risk miRNAs via the regulation of the pivotal risk genes MAPK1, AKT1, and VEGFA. Among them, VEGFA was validated to be significantly decreased in the CSF cells of MS patients by transcriptomic datasets. These findings may provide potential biomarkers or therapeutic targets and help elucidate the molecular mechanisms underlying the pathogenesis of MS.
期刊介绍:
The Journal of Molecular Neuroscience is committed to the rapid publication of original findings that increase our understanding of the molecular structure, function, and development of the nervous system. The criteria for acceptance of manuscripts will be scientific excellence, originality, and relevance to the field of molecular neuroscience. Manuscripts with clinical relevance are especially encouraged since the journal seeks to provide a means for accelerating the progression of basic research findings toward clinical utilization. All experiments described in the Journal of Molecular Neuroscience that involve the use of animal or human subjects must have been approved by the appropriate institutional review committee and conform to accepted ethical standards.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


