{"title":"PM2.5 air pollutant drives the initiate of lung adenocarcinoma","authors":"Yuhong Xu, Huiyan Luo","doi":"10.1002/mef2.53","DOIUrl":null,"url":null,"abstract":"<p>Recently, researchers from Cancer Research UK and The Francis Crick Institute published a paper entitled “Lung adenocarcinoma promotion by air pollutants” in Nature.<span><sup>1</sup></span> The study focused on the impact of air pollutants, specifically PM2.5, on lung adenocarcinoma development. By analyzing human data and conducting subsequent animal experiments, the researchers found that air pollutants PM2.5 leads to an influx of macrophages into the lung and triggers the release of interleukin-1β. This, in turn, induces a progenitor-like cell state within estimated glomerular filtration rate (EGFR) mutant lung alveolar type II epithelial cells, fueling tumorigenesis, and potentially exacerbating pre-existing cancerous mutations in normal tissues.</p><p>While the association between smoking and lung cancer risk is well-established, attention has increasingly turned towards understanding the carcinogenic factors in never-smokers. As the eighth leading cause of cancer-related deaths in the United Kingdom, lung cancer in never-smokers (LCINS) is often an adenocarcinoma carrying the EGFR mutation.<span><sup>2</sup></span> In an effort to identify significant factors influencing the development of lung cancer LCINS, the researchers analyzed environmental and epidemiological data from 32,957 cases of EGFR-driven lung cancer in the United Kingdom, Canada, South Korea, Taiwan, and China. The findings revealed a correlation between increased levels of PM2.5 and a higher incidence of lung cancer among the study participants. Later analysis of 407,509 individuals from the UK Biobank support these results, demonstrating significant increase in the projected incidence of lung cancer among those exposed to high levels of PM2.5. The researchers also conducted a 3-year follow-up study involving 228 Canadian lung cancer patients. The incidence of lung cancer was found to be significantly higher (73%) in those exposed to high levels of PM2.5 compared to those exposed to low levels (40%). Notably, this association was not observed in the Canadian cohort over a 20-year period, suggesting that 3 years of exposure to high levels of pollution may be sufficient to produce cancer.</p><p>Hill et al. further employed genetically engineered mice carrying EGFR mutations (EGFR<sup>L858R</sup>) associated with human cancer to functionally investigate whether PM2.5 exposure promoted the development of lung adenocarcinoma. The study revealed that mice were exposed to similar air pollution particles, resulting in a higher likelihood of developing lung tumors compared to control mice not exposed to pollution particles. The same experiments were performed on genetically engineered mice with Kras mutations, a common mutation in various lung tumors, yielding similar results. Through spatial analysis of clonal dynamics, the researchers discovered that PM2.5 promotes early tumorigenesis through two mechanisms: increasing the number of EGFR-mutated cells capable of forming tumors and enhancing the proliferation rate of these mutated cells in early lesions. To determine whether PM2.5 induces DNA mutations, the researchers conducted whole-genome sequencing of tumors from EGFR<sup>L858R</sup> mice exposed to PM2.5 or a control substance (phosphate buffered saline [PBS]). The results suggested that short-term PM2.5 exposure does not enhance mutations, and PM2.5-induced lung tumorigenesis driven by EGFR requires a functional immune system. Inhalation of toxic particles triggers a local response in the lungs, mediated by macrophages and lung epithelial cells. Transient exposure to PM2.5 was found to be associated with increased and sustained infiltration of pulmonary macrophages after the exposure period. Additionally, to investigate the effects of PM2.5 exposure on early tumorigenesis, researchers used RNA-seq analysis of lung epithelial cells in four different conditions (control mice exposed to PM2.5 or PBS, and mice harboring an EGFR mutation exposed to PM2.5 or PBS) revealed that the IL-6–JAK–STAT pathway, inflammatory responses and the allograft rejection pathway was upregulated in the PM2.5 exposure group compared to the control group. PM2.5 exposure alse led to upregulation of genes involved in macrophage recruitment, including genes encoding interleukin-1β (IL-1β), GM-CSF, CCL6, and NF-κB and the epithelial-derived alarmin IL-33.</p><p>Based on previous studies suggesting that alveolar type II (AT2) epithelial cells may be a source of lung adenocarcinoma,<span><sup>3</sup></span> Nagano et al. compared bulk RNA-seq expression data and a single-cell RNA-seq data set of bleomycin-treated mouse lungs. The analysis indicated that the activation score of AT2 progenitor cells was higher in the PM2.5 exposure group compared to the control group. This suggests that, in the presence of an EGFR mutation and PM2.5 exposure, AT2 cells undergo transcriptional reprogramming, transitioning into a progenitor cell state. Importantly, this effect was observed solely in EGFR<sup>L858R</sup> AT2 cells, and not in EGFR wild-type AT2 cells. Furthermore, a comparison of mouse RNA-seq data with human clinical cross-over studies revealed the upregulation of many genes in mice lung epithelium that were also upregulated in human lung epithelium upon PM2.5 exposure.</p><p>Previous studies have shown that PM2.5 exposure can increase the release of inflammatory cytokines from macrophages.<span><sup>4</sup></span> By coculture AT2 cells from EGFR<sup>L858R</sup> mice with macrophages exposed to PM2.5 or PBS, the researchers observed a significantly increased organoid-formation efficiency of AT2 cells in the PM2.5 exposure group. This indicated that the key mediators of PM2.5-induced inflammation originate from macrophages. Earlier reports have highlighted the requirement of IL-1β in lung macrophages for AT2 progenitor cell formation.<span><sup>5</sup></span> By combining existing data, the researchers concluded that, upon exposure to fine particulate matter, lung epithelial cells recruit macrophages into the lungs. Fine particulate matter then stimulates macrophages to release IL-1β, leading to the reprogramming of EGFR<sup>L858R</sup> AT2 into a progenitor cell state which subsequently becomes the seed for initiating lung cancer (Figure 1). The researchers further validated these conclusions through IL-1β antibody treatment experiment during PM2.5 exposure.</p><p>To get preliminary insights into the prevalence of EGFR or Kras mutations among individuals, the researchers analyzed surveillance data from different cohorts. Among 295 healthy lung tissue samples, 54 (18%) were found to carry the EGFR-driving mutation. Similarly, among 81 healthy lung tissue samples, 43 (53%) were found to carry Kras-driving mutations. Notably, only one in 554,500 healthy lung cells was found to carry a carcinogenic EGFR mutation. Furthermore, a significant correlation was observed between age and the number of mutations, while no association was found between EGFR or Kras mutations and smoking status or cancer diagnosis in noncancerous tissues.</p><p>This research has demonstrated the existence of cancer-causing mutations in healthy tissues and highlighted the ability of normal cells, which undergo spontaneous genetic mutations during proliferation, to transform into malignant cells and initiate cancer under the influence of external environment such as PM2.5. And PM2.5 is one of the possible risk factors for the development of lung adenocarcinoma, and the influence of the lung immune system may be the key to its role. Consequently, public health initiatives aimed at reducing air pollution have the potential to effectively mitigate the burden of lung cancer. Moreover, these findings have implications for cancer prevention, suggesting that anti-inflammatory interventions may prevent the development of such cancers.</p><p><b>Yuhong Xu</b>: Visualization (equal); writing—original draft (equal); writing—review and editing (equal). <b>Huiyan Luo</b>: Conceptualization (equal); funding acquisition (equal); supervision (equal); writing—review and editing (equal). Both authors have read and approved the article.</p><p>The authors declare no conflict of interest.</p><p>Not applicable.</p>","PeriodicalId":74135,"journal":{"name":"MedComm - Future medicine","volume":"2 3","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2023-07-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mef2.53","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm - Future medicine","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mef2.53","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Recently, researchers from Cancer Research UK and The Francis Crick Institute published a paper entitled “Lung adenocarcinoma promotion by air pollutants” in Nature.1 The study focused on the impact of air pollutants, specifically PM2.5, on lung adenocarcinoma development. By analyzing human data and conducting subsequent animal experiments, the researchers found that air pollutants PM2.5 leads to an influx of macrophages into the lung and triggers the release of interleukin-1β. This, in turn, induces a progenitor-like cell state within estimated glomerular filtration rate (EGFR) mutant lung alveolar type II epithelial cells, fueling tumorigenesis, and potentially exacerbating pre-existing cancerous mutations in normal tissues.
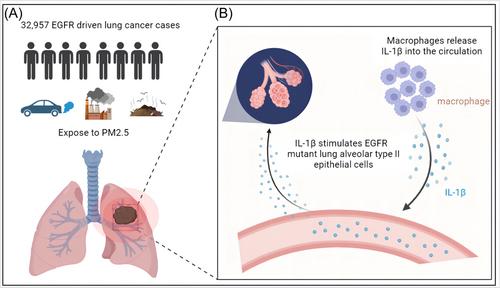
While the association between smoking and lung cancer risk is well-established, attention has increasingly turned towards understanding the carcinogenic factors in never-smokers. As the eighth leading cause of cancer-related deaths in the United Kingdom, lung cancer in never-smokers (LCINS) is often an adenocarcinoma carrying the EGFR mutation.2 In an effort to identify significant factors influencing the development of lung cancer LCINS, the researchers analyzed environmental and epidemiological data from 32,957 cases of EGFR-driven lung cancer in the United Kingdom, Canada, South Korea, Taiwan, and China. The findings revealed a correlation between increased levels of PM2.5 and a higher incidence of lung cancer among the study participants. Later analysis of 407,509 individuals from the UK Biobank support these results, demonstrating significant increase in the projected incidence of lung cancer among those exposed to high levels of PM2.5. The researchers also conducted a 3-year follow-up study involving 228 Canadian lung cancer patients. The incidence of lung cancer was found to be significantly higher (73%) in those exposed to high levels of PM2.5 compared to those exposed to low levels (40%). Notably, this association was not observed in the Canadian cohort over a 20-year period, suggesting that 3 years of exposure to high levels of pollution may be sufficient to produce cancer.
Hill et al. further employed genetically engineered mice carrying EGFR mutations (EGFRL858R) associated with human cancer to functionally investigate whether PM2.5 exposure promoted the development of lung adenocarcinoma. The study revealed that mice were exposed to similar air pollution particles, resulting in a higher likelihood of developing lung tumors compared to control mice not exposed to pollution particles. The same experiments were performed on genetically engineered mice with Kras mutations, a common mutation in various lung tumors, yielding similar results. Through spatial analysis of clonal dynamics, the researchers discovered that PM2.5 promotes early tumorigenesis through two mechanisms: increasing the number of EGFR-mutated cells capable of forming tumors and enhancing the proliferation rate of these mutated cells in early lesions. To determine whether PM2.5 induces DNA mutations, the researchers conducted whole-genome sequencing of tumors from EGFRL858R mice exposed to PM2.5 or a control substance (phosphate buffered saline [PBS]). The results suggested that short-term PM2.5 exposure does not enhance mutations, and PM2.5-induced lung tumorigenesis driven by EGFR requires a functional immune system. Inhalation of toxic particles triggers a local response in the lungs, mediated by macrophages and lung epithelial cells. Transient exposure to PM2.5 was found to be associated with increased and sustained infiltration of pulmonary macrophages after the exposure period. Additionally, to investigate the effects of PM2.5 exposure on early tumorigenesis, researchers used RNA-seq analysis of lung epithelial cells in four different conditions (control mice exposed to PM2.5 or PBS, and mice harboring an EGFR mutation exposed to PM2.5 or PBS) revealed that the IL-6–JAK–STAT pathway, inflammatory responses and the allograft rejection pathway was upregulated in the PM2.5 exposure group compared to the control group. PM2.5 exposure alse led to upregulation of genes involved in macrophage recruitment, including genes encoding interleukin-1β (IL-1β), GM-CSF, CCL6, and NF-κB and the epithelial-derived alarmin IL-33.
Based on previous studies suggesting that alveolar type II (AT2) epithelial cells may be a source of lung adenocarcinoma,3 Nagano et al. compared bulk RNA-seq expression data and a single-cell RNA-seq data set of bleomycin-treated mouse lungs. The analysis indicated that the activation score of AT2 progenitor cells was higher in the PM2.5 exposure group compared to the control group. This suggests that, in the presence of an EGFR mutation and PM2.5 exposure, AT2 cells undergo transcriptional reprogramming, transitioning into a progenitor cell state. Importantly, this effect was observed solely in EGFRL858R AT2 cells, and not in EGFR wild-type AT2 cells. Furthermore, a comparison of mouse RNA-seq data with human clinical cross-over studies revealed the upregulation of many genes in mice lung epithelium that were also upregulated in human lung epithelium upon PM2.5 exposure.
Previous studies have shown that PM2.5 exposure can increase the release of inflammatory cytokines from macrophages.4 By coculture AT2 cells from EGFRL858R mice with macrophages exposed to PM2.5 or PBS, the researchers observed a significantly increased organoid-formation efficiency of AT2 cells in the PM2.5 exposure group. This indicated that the key mediators of PM2.5-induced inflammation originate from macrophages. Earlier reports have highlighted the requirement of IL-1β in lung macrophages for AT2 progenitor cell formation.5 By combining existing data, the researchers concluded that, upon exposure to fine particulate matter, lung epithelial cells recruit macrophages into the lungs. Fine particulate matter then stimulates macrophages to release IL-1β, leading to the reprogramming of EGFRL858R AT2 into a progenitor cell state which subsequently becomes the seed for initiating lung cancer (Figure 1). The researchers further validated these conclusions through IL-1β antibody treatment experiment during PM2.5 exposure.
To get preliminary insights into the prevalence of EGFR or Kras mutations among individuals, the researchers analyzed surveillance data from different cohorts. Among 295 healthy lung tissue samples, 54 (18%) were found to carry the EGFR-driving mutation. Similarly, among 81 healthy lung tissue samples, 43 (53%) were found to carry Kras-driving mutations. Notably, only one in 554,500 healthy lung cells was found to carry a carcinogenic EGFR mutation. Furthermore, a significant correlation was observed between age and the number of mutations, while no association was found between EGFR or Kras mutations and smoking status or cancer diagnosis in noncancerous tissues.
This research has demonstrated the existence of cancer-causing mutations in healthy tissues and highlighted the ability of normal cells, which undergo spontaneous genetic mutations during proliferation, to transform into malignant cells and initiate cancer under the influence of external environment such as PM2.5. And PM2.5 is one of the possible risk factors for the development of lung adenocarcinoma, and the influence of the lung immune system may be the key to its role. Consequently, public health initiatives aimed at reducing air pollution have the potential to effectively mitigate the burden of lung cancer. Moreover, these findings have implications for cancer prevention, suggesting that anti-inflammatory interventions may prevent the development of such cancers.
Yuhong Xu: Visualization (equal); writing—original draft (equal); writing—review and editing (equal). Huiyan Luo: Conceptualization (equal); funding acquisition (equal); supervision (equal); writing—review and editing (equal). Both authors have read and approved the article.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


