A transferable recommender approach for selecting the best density functional approximations in chemical discovery
IF 18.3
Q1 COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS
引用次数: 5
Abstract
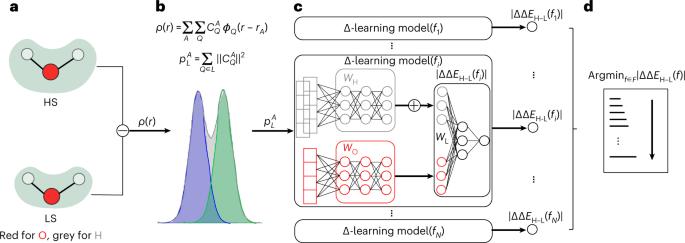
Approximate density functional theory has become indispensable owing to its balanced cost–accuracy trade-off, including in large-scale screening. To date, however, no density functional approximation (DFA) with universal accuracy has been identified, leading to uncertainty in the quality of data generated from density functional theory. With electron density fitting and Δ-learning, we build a DFA recommender that selects the DFA with the lowest expected error with respect to the gold standard (but cost-prohibitive) coupled cluster theory in a system-specific manner. We demonstrate this recommender approach on the evaluation of vertical spin splitting energies of transition metal complexes. Our recommender predicts top-performing DFAs and yields excellent accuracy (about 2 kcal mol−1) for chemical discovery, outperforming both individual Δ-learning models and the best conventional single-functional approach from a set of 48 DFAs. By demonstrating transferability to diverse synthesized compounds, our recommender potentially addresses the accuracy versus scope dilemma broadly encountered in computational chemistry. A density functional recommender enables chemical space exploration by selecting the best exchange–correlation functional for each system, outperforming the use of a single functional for all systems or transfer learning models.
化学发现中选择最佳密度泛函近似的可转移推荐方法
近似密度泛函理论在成本与精确度之间权衡利弊,已成为大规模筛选等研究中不可或缺的方法。然而,迄今为止,尚未发现具有普遍准确性的密度泛函近似(DFA),导致密度泛函理论生成的数据质量不确定。通过电子密度拟合和Δ学习,我们建立了一种密度泛函近似推荐器,它能以特定系统的方式选择与黄金标准(但成本高昂)耦合簇理论相比预期误差最小的密度泛函近似。我们在评估过渡金属复合物的垂直自旋分裂能时演示了这种推荐方法。我们的推荐器预测了性能最佳的 DFA,并为化学发现提供了极高的准确度(约 2 kcal mol-1),其性能优于单独的 Δ 学习模型和来自 48 个 DFA 的最佳传统单功能方法。我们的推荐器能适用于多种合成化合物,从而有可能解决计算化学中普遍存在的精度与范围的两难问题。密度函数推荐器通过为每个系统选择最佳交换相关函数来实现化学空间探索,优于对所有系统或迁移学习模型使用单一函数的方法。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


