Alexander V. Popov, Anton V. Endutkin, Yuri N. Vorobjev, Dmitry O. Zharkov
{"title":"Molecular dynamics simulation of the opposite-base preference and interactions in the active site of formamidopyrimidine-DNA glycosylase","authors":"Alexander V. Popov, Anton V. Endutkin, Yuri N. Vorobjev, Dmitry O. Zharkov","doi":"10.1186/s12900-017-0075-y","DOIUrl":null,"url":null,"abstract":"<p>Formamidopyrimidine-DNA glycosylase (Fpg) removes abundant pre-mutagenic 8-oxoguanine (oxoG) bases from DNA through nucleophilic attack of its N-terminal proline at C1′ of the damaged nucleotide. Since oxoG efficiently pairs with both C and A, Fpg must excise oxoG from pairs with C but not with A, otherwise a mutation occurs. The crystal structures of several Fpg–DNA complexes have been solved, yet no structure with A opposite the lesion is available.</p><p>Here we use molecular dynamic simulation to model interactions in the pre-catalytic complex of <i>Lactococcus lactis</i> Fpg with DNA containing oxoG opposite C or A, the latter in either <i>syn</i> or <i>anti</i> conformation. The catalytic dyad, Pro1–Glu2, was modeled in all four possible protonation states. Only one transition was observed in the experimental reaction rate pH dependence plots, and Glu2 kept the same set of interactions regardless of its protonation state, suggesting that it does not limit the reaction rate. The adenine base opposite oxoG was highly distorting for the adjacent nucleotides: in the more stable <i>syn</i> models it formed non-canonical bonds with out-of-register nucleotides in both the damaged and the complementary strand, whereas in the <i>anti</i> models the adenine either formed non-canonical bonds or was expelled into the major groove. The side chains of Arg109 and Phe111 that Fpg inserts into DNA to maintain its kinked conformation tended to withdraw from their positions if A was opposite to the lesion. The region showing the largest differences in the dynamics between oxoG:C and oxoG:A substrates was unexpectedly remote from the active site, located near the linker joining the two domains of Fpg. This region was also highly conserved among 124 analyzed Fpg sequences. Three sites trapping water molecules through multiple bonds were identified on the protein–DNA interface, apparently helping to maintain enzyme-induced DNA distortion and participating in oxoG recognition.</p><p>Overall, the discrimination against A opposite to the lesion seems to be due to incorrect DNA distortion around the lesion-containing base pair and, possibly, to gross movement of protein domains connected by the linker.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"17 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2017-05-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-017-0075-y","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-017-0075-y","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 4
Abstract
Formamidopyrimidine-DNA glycosylase (Fpg) removes abundant pre-mutagenic 8-oxoguanine (oxoG) bases from DNA through nucleophilic attack of its N-terminal proline at C1′ of the damaged nucleotide. Since oxoG efficiently pairs with both C and A, Fpg must excise oxoG from pairs with C but not with A, otherwise a mutation occurs. The crystal structures of several Fpg–DNA complexes have been solved, yet no structure with A opposite the lesion is available.
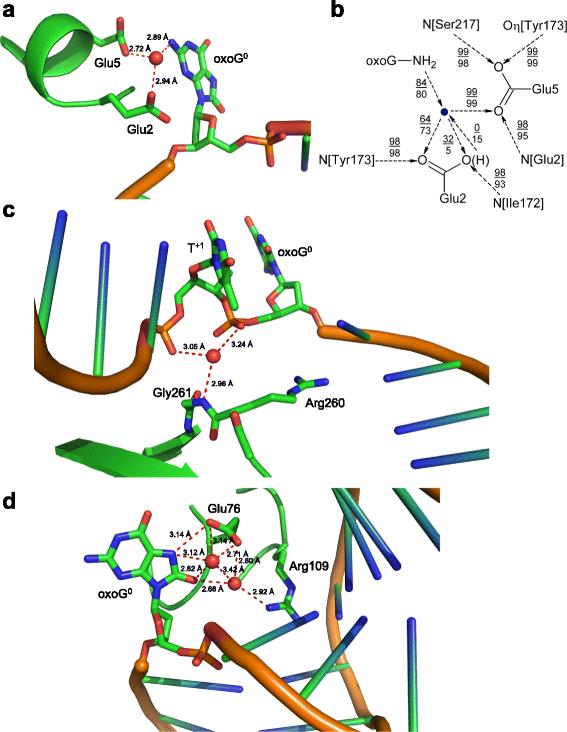
Here we use molecular dynamic simulation to model interactions in the pre-catalytic complex of Lactococcus lactis Fpg with DNA containing oxoG opposite C or A, the latter in either syn or anti conformation. The catalytic dyad, Pro1–Glu2, was modeled in all four possible protonation states. Only one transition was observed in the experimental reaction rate pH dependence plots, and Glu2 kept the same set of interactions regardless of its protonation state, suggesting that it does not limit the reaction rate. The adenine base opposite oxoG was highly distorting for the adjacent nucleotides: in the more stable syn models it formed non-canonical bonds with out-of-register nucleotides in both the damaged and the complementary strand, whereas in the anti models the adenine either formed non-canonical bonds or was expelled into the major groove. The side chains of Arg109 and Phe111 that Fpg inserts into DNA to maintain its kinked conformation tended to withdraw from their positions if A was opposite to the lesion. The region showing the largest differences in the dynamics between oxoG:C and oxoG:A substrates was unexpectedly remote from the active site, located near the linker joining the two domains of Fpg. This region was also highly conserved among 124 analyzed Fpg sequences. Three sites trapping water molecules through multiple bonds were identified on the protein–DNA interface, apparently helping to maintain enzyme-induced DNA distortion and participating in oxoG recognition.
Overall, the discrimination against A opposite to the lesion seems to be due to incorrect DNA distortion around the lesion-containing base pair and, possibly, to gross movement of protein domains connected by the linker.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


