{"title":"Impact of genome and epigenome on intratumor heterogeneity in colorectal cancer","authors":"Jia-qian Huang, Hui-yan Luo","doi":"10.1002/mef2.34","DOIUrl":null,"url":null,"abstract":"<p>Recently, two companion papers published in <i>Nature</i> by Timon Heide et al.<span><sup>1</sup></span> and Jacob Househam et al.<span><sup>2</sup></span> suggested that phenotypic characteristics can vary without heritable (epi)genetic alteration to drive gene expression change, namely phenotypic plastic, which could take part in intratumor heterogeneity in colorectal cancer (CRC) evolution.</p><p>As in other cancer types, intratumor heterogeneity represents a challenge in the facets of tumorigenesis, evolution, and therapy response in CRC.<span><sup>3</sup></span> However, the information on how genomes and epigenomes contribute to intratumor heterogeneity is limited. What's more, although consensus molecular subtypes (CMS) and CRC intrinsic subtypes (CRIS) are approaches used to classify CRC cases by gene expression patterns,<span><sup>4</sup></span> Househam et al.<span><sup>2</sup></span> found that very few tumors with sufficient samples could be homogeneously classified by these classifiers, indicating intratumor heterogeneity of molecular subtypes in CRC and the discrepancy between CMS and CRIS classifications. Therefore, the researchers collected a large number of samples from a multiregion of carcinoma, concomitant adenoma if present, and a distant region of the normal epithelium to integrate their spatially resolved mutiomics analysis with single gland profiling data set, and combine with computational modeling to understand the cancer cell biology and assess the functional impact of altered gene expression on the evolution of CRC, due to intratumor heterogeneity is a significant confounding factor in bulk-tumor profiling (Figure 1). As for spatially resolved multiomics analysis, it presents a new avenue to reveal tumors and microenvironments co-evolution, which could be used to clarify heterogeneity.<span><sup>5</sup></span> In detail, it contains a strategy for the spatial sampling of tumor tissue to implement a series of new spatial genomic, transcriptomic, and proteomic technologies.<span><sup>5</sup></span></p><p>Heide et al.<span><sup>1</sup></span> first looked forward to measuring genome–epigenome co-evaluation in a quantitative manner and gained some evidence. First of all, it was confirmed that there were recurrent cancer driver mutation events in CRC. After examining somatic mutations in chromatin modifier genes and assessing the evolutionary selection by the ratio of nonsynonymous to synonymous substitutions (<i>dN</i>/<i>dS</i>), they identified clear evidence of clonal truncating mutations of chromatin modifiers genes, which indicated that somatic mutations affect the epigenome. Besides, somatic chromatin accessibility alterations (SCAAs), which were found in known driver genes without accompanying mutations, were a substitute pattern for driver gene (in)activation. Subsequently, Heide et al. found that most SCAAs occurred at the onset of the adenoma-carcinoma transition. And SCAAs were indeed changes that originated during tumorigenesis instead of the product of normal tissue aging. In addition, SCAAs might change the expression of associated genes.</p><p>What's more, Heide et al. detected genome-wide differential chromatin accessibility alteration of transcription factor-binding sites (including the interferon-regulatory factor family, the CCCTC-binding factor, and the HOX, FOX, and SOX families) in CRC, which was reported that some of these binding sites were demethylated.<span><sup>1</sup></span> And the alteration was stable and heritable. Taken together, the study utilized spatially resolved multiomics analysis to figure out the nongenetic determinants of cancer cell biology and clonal evolution in CRC. To explore whether the variability of gene expression resulted from genetic change, Househam et al. constructed phylogenetic trees for each tumor. However, only 61 out of 8368 genes recurrently reflected phylogenetic ancestry, and only the peroxisome proliferator-activated receptor signaling pathway was significantly shown with recurrent evidence of phylogenetic signal. Therefore, further study was on the influence of tumor microenvironment (TME) and discovered that TME could affect plastic gene expression programs irrespective of accrued genetic change, while Heide et al.<span><sup>1</sup></span> demonstrated that the epigenome, in turn, contributes to the accumulation of DNA mutations.</p><p>Given that phylogenetic signals do exist, Househam et al. then presented a linear regression framework to examine <i>cis</i>-associations between inter- and intratumor somatic genetic heterogeneity and gene expression and found 5927 genes ultimately, since gene expression could be modified by somatic mutations is a latent mechanistic interpretation. They measured that 1529 out of 5927 (25.8%) genes had gene expression associated with somatic genetic variations, which they termed expression quantitative trait loci (eQTL) genes. Despite a large proportion of somatic mutations having little influence on <i>cis</i> gene expression, there remained 2.4% of subclonal genetic variants related with altered gene expression and they were enriched for phylogenetic signal.</p><p>Secondly, Househam et al. focused on investigating the function of drivers that were considered to foster cancer evolution and their mutations. To accurately identify clone and subclone somatic variants and to call somatic copy number alterations, they used their extensive single-gland, multiregion whole genome sequencing (WGS) data, and low-pass WGS data. Most of the frequently mutated genes in CRC were clonal in their cases, except for two of them that had subclone <i>KRAS</i> or <i>TP53</i> mutation. Thus, <i>dN</i>/<i>dS</i> was used to detect the selection of driver genes again. The ratio was greater than 1 for subclone variants in microsatellite stability, showing evidence of positive selection of a subset of putative subclonal CRC driver mutations in growing tumors, but not in microsatellite instability, which is similar to the result from Heide et al.<span><sup>1</sup></span> Using DepMap data set for implementation of orthogonal assessment, seldom putative drivers demonstrated evidence of essentiality in CRC cell lines. Overall, based on the selection of subclones, even driver mutations played a slight role in phenotypic consequences.</p><p>Subsequently, Househam et al. discovered balanced status in the majority of tumors, which indicates analogous branch lengths across samples and regions from the same tumor, when assessing the evolutionary dynamics via evaluation of the phylogenetic tree shape and the related clonal structure of the tumor. Given an “unbalanced” tree shown from tumor C539, BaseScope was used to visualize subclones and found the spatial segregation of subclones, which suggests that a subset of the blocks is heterogeneous. To identify the subclone variants previously selected by <i>dN/dS</i>, Househam et al. designed a spatial inference framework based on Approximate Bayesian Computation-Sequential Monte Carlo (ABC-SMC) to achieve computational modeling. To compare the model with authentic data, they simulated the sampling scheme on virtual tumors and reconstructed a phylogenetic tree, whose structures were generally consistent with the observed phylogenetic tree. Significant evidence of subclone selection was present in 7 out of 27 tumors; in addition, 4 of the 7 tumors presented a putative subclone mutation, and the variant was expressed in the RNA. Moreover, tumors were characterized by exponentially growing or growing more slowly at the periphery exclusively, and the growth rate of subclones that underwent selection was 20-fold higher than the background clone, and most of them originated in the early stages of tumor expansion.</p><p>Finally, Househam et al. wondered about the reason for the evolution of the selected subclones. Examining matched transposase-accessible chromatin sequencing (ATAC-seq) and RNA-seq from selected subclones to generate epigenome and transcriptome. Dysregulation of focal adhesion pathways, upregulation of the epithelial-mesenchymal transition program that was confirmed by Heide et al.,<span><sup>1</sup></span> and upregulation of MYC + E2F targets was found through enrichment analysis of differentially expressed genes between the subclone and background clone. Moreover, there were no proof that heritable variations in gene expression were able to suggest subclone selection, hinting that transcriptional variation remains occurring even in a selected clone.</p><p>Herein, we have recognized the significance of epigenome for CRC evolution through these companion papers. Much work is still needed to better understand the critical role of epigenetic signatures in cancer initiation and progression, including further functional study and clarification of the mechanistic link. Nevertheless, these studies uncovered another factor, epigenetics, which universally influenced phenotype on cancer cells, and provided a perspective to better understand heterogeneity in CRC.</p><p><b>Jia-qian Huang</b>: Visualization (lead); writing—original draft (lead); writing—review & editing (equal). <b>Hui-yan Luo</b>: Conceptualization (lead); funding acquisition (lead); supervision (lead); writing—review & editing (equal). Both authors have read and approved the article.</p><p>Not applicable.</p><p>Not applicable.</p>","PeriodicalId":74135,"journal":{"name":"MedComm - Future medicine","volume":"2 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2023-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mef2.34","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm - Future medicine","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mef2.34","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Recently, two companion papers published in Nature by Timon Heide et al.1 and Jacob Househam et al.2 suggested that phenotypic characteristics can vary without heritable (epi)genetic alteration to drive gene expression change, namely phenotypic plastic, which could take part in intratumor heterogeneity in colorectal cancer (CRC) evolution.
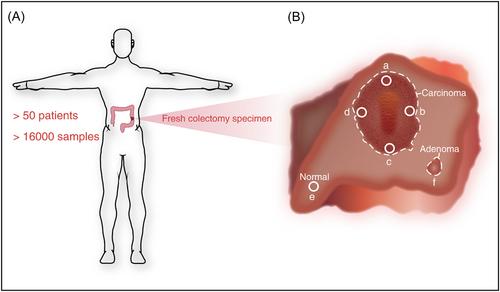
As in other cancer types, intratumor heterogeneity represents a challenge in the facets of tumorigenesis, evolution, and therapy response in CRC.3 However, the information on how genomes and epigenomes contribute to intratumor heterogeneity is limited. What's more, although consensus molecular subtypes (CMS) and CRC intrinsic subtypes (CRIS) are approaches used to classify CRC cases by gene expression patterns,4 Househam et al.2 found that very few tumors with sufficient samples could be homogeneously classified by these classifiers, indicating intratumor heterogeneity of molecular subtypes in CRC and the discrepancy between CMS and CRIS classifications. Therefore, the researchers collected a large number of samples from a multiregion of carcinoma, concomitant adenoma if present, and a distant region of the normal epithelium to integrate their spatially resolved mutiomics analysis with single gland profiling data set, and combine with computational modeling to understand the cancer cell biology and assess the functional impact of altered gene expression on the evolution of CRC, due to intratumor heterogeneity is a significant confounding factor in bulk-tumor profiling (Figure 1). As for spatially resolved multiomics analysis, it presents a new avenue to reveal tumors and microenvironments co-evolution, which could be used to clarify heterogeneity.5 In detail, it contains a strategy for the spatial sampling of tumor tissue to implement a series of new spatial genomic, transcriptomic, and proteomic technologies.5
Heide et al.1 first looked forward to measuring genome–epigenome co-evaluation in a quantitative manner and gained some evidence. First of all, it was confirmed that there were recurrent cancer driver mutation events in CRC. After examining somatic mutations in chromatin modifier genes and assessing the evolutionary selection by the ratio of nonsynonymous to synonymous substitutions (dN/dS), they identified clear evidence of clonal truncating mutations of chromatin modifiers genes, which indicated that somatic mutations affect the epigenome. Besides, somatic chromatin accessibility alterations (SCAAs), which were found in known driver genes without accompanying mutations, were a substitute pattern for driver gene (in)activation. Subsequently, Heide et al. found that most SCAAs occurred at the onset of the adenoma-carcinoma transition. And SCAAs were indeed changes that originated during tumorigenesis instead of the product of normal tissue aging. In addition, SCAAs might change the expression of associated genes.
What's more, Heide et al. detected genome-wide differential chromatin accessibility alteration of transcription factor-binding sites (including the interferon-regulatory factor family, the CCCTC-binding factor, and the HOX, FOX, and SOX families) in CRC, which was reported that some of these binding sites were demethylated.1 And the alteration was stable and heritable. Taken together, the study utilized spatially resolved multiomics analysis to figure out the nongenetic determinants of cancer cell biology and clonal evolution in CRC. To explore whether the variability of gene expression resulted from genetic change, Househam et al. constructed phylogenetic trees for each tumor. However, only 61 out of 8368 genes recurrently reflected phylogenetic ancestry, and only the peroxisome proliferator-activated receptor signaling pathway was significantly shown with recurrent evidence of phylogenetic signal. Therefore, further study was on the influence of tumor microenvironment (TME) and discovered that TME could affect plastic gene expression programs irrespective of accrued genetic change, while Heide et al.1 demonstrated that the epigenome, in turn, contributes to the accumulation of DNA mutations.
Given that phylogenetic signals do exist, Househam et al. then presented a linear regression framework to examine cis-associations between inter- and intratumor somatic genetic heterogeneity and gene expression and found 5927 genes ultimately, since gene expression could be modified by somatic mutations is a latent mechanistic interpretation. They measured that 1529 out of 5927 (25.8%) genes had gene expression associated with somatic genetic variations, which they termed expression quantitative trait loci (eQTL) genes. Despite a large proportion of somatic mutations having little influence on cis gene expression, there remained 2.4% of subclonal genetic variants related with altered gene expression and they were enriched for phylogenetic signal.
Secondly, Househam et al. focused on investigating the function of drivers that were considered to foster cancer evolution and their mutations. To accurately identify clone and subclone somatic variants and to call somatic copy number alterations, they used their extensive single-gland, multiregion whole genome sequencing (WGS) data, and low-pass WGS data. Most of the frequently mutated genes in CRC were clonal in their cases, except for two of them that had subclone KRAS or TP53 mutation. Thus, dN/dS was used to detect the selection of driver genes again. The ratio was greater than 1 for subclone variants in microsatellite stability, showing evidence of positive selection of a subset of putative subclonal CRC driver mutations in growing tumors, but not in microsatellite instability, which is similar to the result from Heide et al.1 Using DepMap data set for implementation of orthogonal assessment, seldom putative drivers demonstrated evidence of essentiality in CRC cell lines. Overall, based on the selection of subclones, even driver mutations played a slight role in phenotypic consequences.
Subsequently, Househam et al. discovered balanced status in the majority of tumors, which indicates analogous branch lengths across samples and regions from the same tumor, when assessing the evolutionary dynamics via evaluation of the phylogenetic tree shape and the related clonal structure of the tumor. Given an “unbalanced” tree shown from tumor C539, BaseScope was used to visualize subclones and found the spatial segregation of subclones, which suggests that a subset of the blocks is heterogeneous. To identify the subclone variants previously selected by dN/dS, Househam et al. designed a spatial inference framework based on Approximate Bayesian Computation-Sequential Monte Carlo (ABC-SMC) to achieve computational modeling. To compare the model with authentic data, they simulated the sampling scheme on virtual tumors and reconstructed a phylogenetic tree, whose structures were generally consistent with the observed phylogenetic tree. Significant evidence of subclone selection was present in 7 out of 27 tumors; in addition, 4 of the 7 tumors presented a putative subclone mutation, and the variant was expressed in the RNA. Moreover, tumors were characterized by exponentially growing or growing more slowly at the periphery exclusively, and the growth rate of subclones that underwent selection was 20-fold higher than the background clone, and most of them originated in the early stages of tumor expansion.
Finally, Househam et al. wondered about the reason for the evolution of the selected subclones. Examining matched transposase-accessible chromatin sequencing (ATAC-seq) and RNA-seq from selected subclones to generate epigenome and transcriptome. Dysregulation of focal adhesion pathways, upregulation of the epithelial-mesenchymal transition program that was confirmed by Heide et al.,1 and upregulation of MYC + E2F targets was found through enrichment analysis of differentially expressed genes between the subclone and background clone. Moreover, there were no proof that heritable variations in gene expression were able to suggest subclone selection, hinting that transcriptional variation remains occurring even in a selected clone.
Herein, we have recognized the significance of epigenome for CRC evolution through these companion papers. Much work is still needed to better understand the critical role of epigenetic signatures in cancer initiation and progression, including further functional study and clarification of the mechanistic link. Nevertheless, these studies uncovered another factor, epigenetics, which universally influenced phenotype on cancer cells, and provided a perspective to better understand heterogeneity in CRC.
Jia-qian Huang: Visualization (lead); writing—original draft (lead); writing—review & editing (equal). Hui-yan Luo: Conceptualization (lead); funding acquisition (lead); supervision (lead); writing—review & editing (equal). Both authors have read and approved the article.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


