{"title":"A case of 46,XY complete gonadal dysgenesis with a novel missense variant in <i>SRY</i>.","authors":"Chisato Narita, Noriyuki Takubo, Manami Sammori, Yuko Matsumura, Kazuhiro Shimura, Rie Ozaki, Hidenori Haruna, Satoshi Narumi, Tomohiro Ishii, Tomonobu Hasegawa, Toshiaki Shimizu","doi":"10.1297/cpe.2023-0032","DOIUrl":null,"url":null,"abstract":"<p><p>Disorders of sex development (DSD) with mild external genital abnormalities may be diagnosed after puberty. Here, we report a case of 46,XY complete gonadal dysgenesis with a novel missense variant in sex-determining region Y (<i>SRY)</i>, diagnosed after primary amenorrhea. A 15-yr-old patient presented to our gynecology department with a chief complaint of amenorrhea. The patient was diagnosed with a 46,XY karyotype, and SRY gene positivity. Gonadotropin levels were high, whereas testosterone levels were low. A pelvic magnetic resonance imaging (MRI) revealed a hypoplastic uterus; however, no gonads could be identified. Laparoscopy revealed bilateral streak gonads, fallopian tube-like structures, and the uterus. The gonads were removed based on the risk of gonadal malignancy. Comprehensive genetic analysis of DSD revealed a previously unreported <i>SRY</i> variant, c.271A>T, p.Ser91Cys, and <i>in silico</i> analysis predicted the variant to be pathogenic. The patient was diagnosed with 46,XY complete gonadal dysgenesis with a novel missense variant in <i>SRY</i>. The patient continued female hormone replacement therapy and experienced breast enlargement and cyclic menstruation. Determining the etiology of DSD can be difficult, causing anxiety in patients and their families. In addition to surgical scrutiny, genetic analysis is important to aid in diagnosis and reassure patients and their families.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":null,"pages":null},"PeriodicalIF":1.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/40/6a/cpe-32-235.PMC10568573.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2023-0032","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/8 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
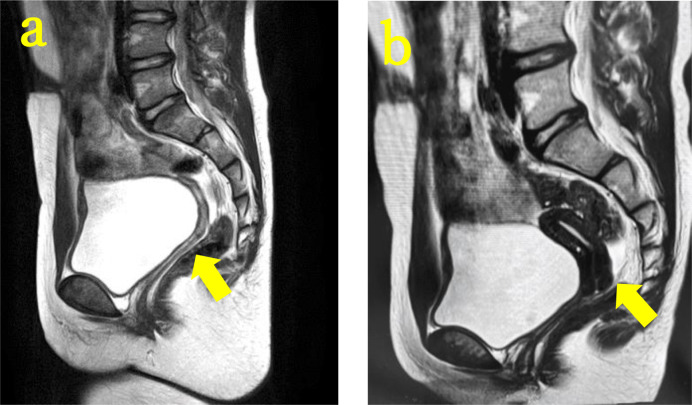
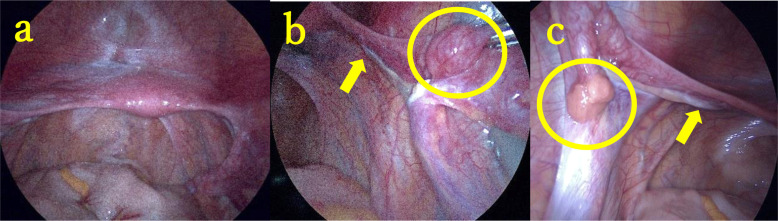
Disorders of sex development (DSD) with mild external genital abnormalities may be diagnosed after puberty. Here, we report a case of 46,XY complete gonadal dysgenesis with a novel missense variant in sex-determining region Y (SRY), diagnosed after primary amenorrhea. A 15-yr-old patient presented to our gynecology department with a chief complaint of amenorrhea. The patient was diagnosed with a 46,XY karyotype, and SRY gene positivity. Gonadotropin levels were high, whereas testosterone levels were low. A pelvic magnetic resonance imaging (MRI) revealed a hypoplastic uterus; however, no gonads could be identified. Laparoscopy revealed bilateral streak gonads, fallopian tube-like structures, and the uterus. The gonads were removed based on the risk of gonadal malignancy. Comprehensive genetic analysis of DSD revealed a previously unreported SRY variant, c.271A>T, p.Ser91Cys, and in silico analysis predicted the variant to be pathogenic. The patient was diagnosed with 46,XY complete gonadal dysgenesis with a novel missense variant in SRY. The patient continued female hormone replacement therapy and experienced breast enlargement and cyclic menstruation. Determining the etiology of DSD can be difficult, causing anxiety in patients and their families. In addition to surgical scrutiny, genetic analysis is important to aid in diagnosis and reassure patients and their families.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


