Benjamin T Enslow, James D Stockand, Jonathan M Berman
{"title":"Liddle's syndrome mechanisms, diagnosis and management.","authors":"Benjamin T Enslow, James D Stockand, Jonathan M Berman","doi":"10.2147/IBPC.S188869","DOIUrl":null,"url":null,"abstract":"<p><p>Liddle's syndrome is a genetic disorder characterized by hypertension with hypokalemic metabolic alkalosis, hyporeninemia and suppressed aldosterone secretion that often appears early in life. It results from inappropriately elevated sodium reabsorption in the distal nephron. Liddle's syndrome is caused by mutations to subunits of the Epithelial Sodium Channel (ENaC). Among other mechanisms, such mutations typically prevent ubiquitination of these subunits, slowing the rate at which they are internalized from the membrane, resulting in an elevation of channel activity. A minority of Liddle's syndrome mutations, though, result in a complementary effect that also elevates activity by increasing the probability that ENaC channels within the membrane are open. Potassium-sparing diuretics such as amiloride and triamterene reduce ENaC activity, and in combination with a reduced sodium diet can restore normotension and electrolyte imbalance in Liddle's syndrome patients and animal models. Liddle's syndrome can be diagnosed clinically by phenotype and confirmed through genetic testing. This review examines the clinical features of Liddle's syndrome, the differential diagnosis of Liddle's syndrome and differentiation from other genetic diseases with similar phenotype, and what is currently known about the population-level prevalence of Liddle's syndrome. This review gives special focus to the molecular mechanisms of Liddle's syndrome.</p>","PeriodicalId":45299,"journal":{"name":"Integrated Blood Pressure Control","volume":"12 ","pages":"13-22"},"PeriodicalIF":2.7000,"publicationDate":"2019-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/46/b1/ibpc-12-13.PMC6731958.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Integrated Blood Pressure Control","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/IBPC.S188869","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"PERIPHERAL VASCULAR DISEASE","Score":null,"Total":0}
引用次数: 0
Abstract
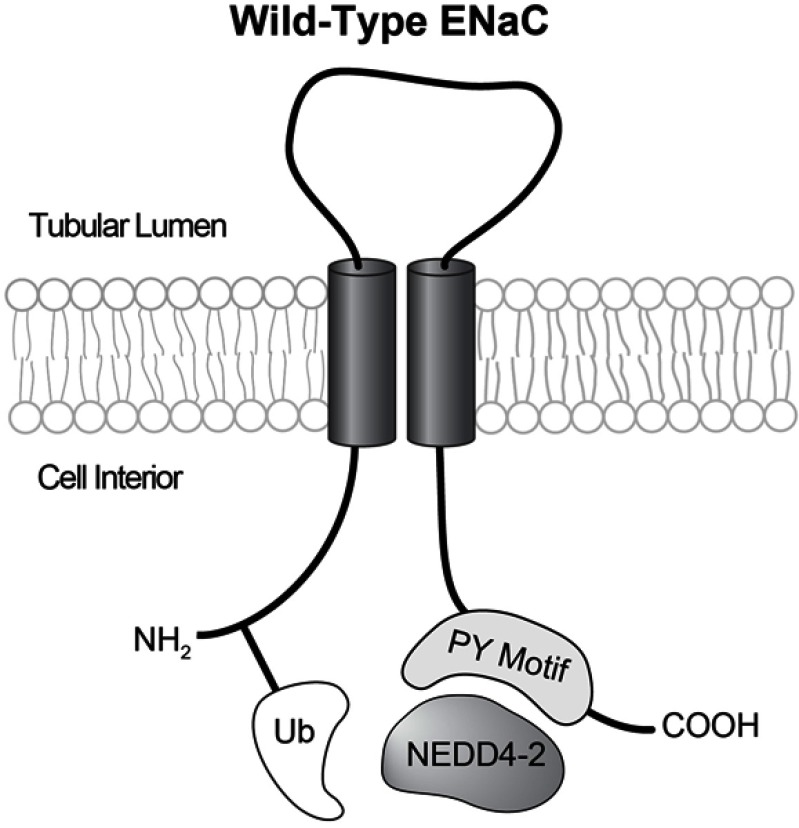
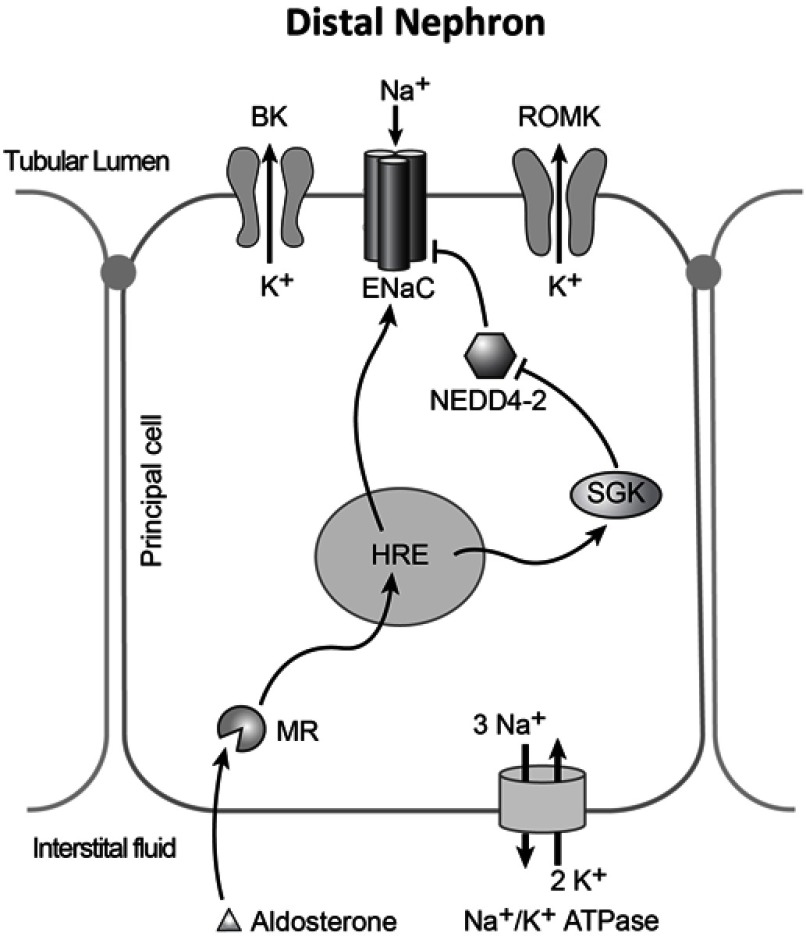
Liddle's syndrome is a genetic disorder characterized by hypertension with hypokalemic metabolic alkalosis, hyporeninemia and suppressed aldosterone secretion that often appears early in life. It results from inappropriately elevated sodium reabsorption in the distal nephron. Liddle's syndrome is caused by mutations to subunits of the Epithelial Sodium Channel (ENaC). Among other mechanisms, such mutations typically prevent ubiquitination of these subunits, slowing the rate at which they are internalized from the membrane, resulting in an elevation of channel activity. A minority of Liddle's syndrome mutations, though, result in a complementary effect that also elevates activity by increasing the probability that ENaC channels within the membrane are open. Potassium-sparing diuretics such as amiloride and triamterene reduce ENaC activity, and in combination with a reduced sodium diet can restore normotension and electrolyte imbalance in Liddle's syndrome patients and animal models. Liddle's syndrome can be diagnosed clinically by phenotype and confirmed through genetic testing. This review examines the clinical features of Liddle's syndrome, the differential diagnosis of Liddle's syndrome and differentiation from other genetic diseases with similar phenotype, and what is currently known about the population-level prevalence of Liddle's syndrome. This review gives special focus to the molecular mechanisms of Liddle's syndrome.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


