{"title":"Genome-wide identification and functional analysis of dysregulated alternative splicing profiles in sepsis.","authors":"Dilixiati Tuerdimaimaiti, Buzukela Abuduaini, Shaotao Kang, Jinliang Jiao, Mengchen Li, Wolazihan Madeniyati, Baihetinisha Tuerdi, Gulisitan Aili, Reyila Tuerhong, Ajiguli Kulaxi","doi":"10.1186/s12950-023-00355-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>An increasing body of evidence now shows that the long-term mortality of patients with sepsis are associated with various sepsis-related immune cell defects. Alternative splicing (AS), as a sepsis-related immune cell defect, is considered as a potential immunomodulatory therapy target to improve patient outcomes. However, our understanding of the role AS plays in sepsis is currently insufficient.</p><p><strong>Aim: </strong>This study investigated possible associations between AS and the gene regulatory networks affecting immune cells. We also investigated apoptosis and AS functionality in sepsis pathophysiology.</p><p><strong>Methods: </strong>In this study, we assessed publicly available mRNA-seq data that was obtained from the NCBI GEO dataset (GSE154918), which included a healthy group (HLTY), a mild infection group (INF1), asepsis group (Seps), and a septic shock group (Shock). A total of 79 samples (excluding significant outliers) were identified by a poly-A capture method to generate RNA-seq data. The variable splicing events and highly correlated RNA binding protein (RBP) genes in each group were then systematically analyzed.</p><p><strong>Results: </strong>For the first time, we used systematic RNA-seq analysis of sepsis-related AS and identified 1505 variable AS events that differed significantly (p <= 0.01) across the four groups. In the sepsis group, the genes related to significant AS events, such as, SHISA5 and IFI27, were mostly enriched in the cell apoptosis pathway. Furthermore, we identified differential splicing patterns within each of the four groups. Significant differences in the expression of RNA Binding Protein(RBP) genes were observed between the control group and the sepsis group. RBP gene expression was highly correlated with variant splicing events in sepsis, as determined by co-expression analysis; The expression of DDX24, CBFA2T2, NOP, ILF3, DNMT1, FTO, PPRC1, NOLC1 RBPs were significant reduced in sepsis compared to the healthy group. Finally, we constructed an RBP-AS functional network.</p><p><strong>Conclusion: </strong>Analysis indicated that the RBP-AS functional network serves as a critical post-transcriptional mechanism that regulates the development of sepsis. AS dysregulation is associated with alterations in the regulatory gene expression network that is involved in sepsis. Therefore, the RBP-AS expression network could be useful in refining biomarker predictions in the development of new therapeutic targets for the pathogenesis of sepsis.</p>","PeriodicalId":56120,"journal":{"name":"Journal of Inflammation-London","volume":"20 1","pages":"31"},"PeriodicalIF":4.4000,"publicationDate":"2023-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10521395/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inflammation-London","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12950-023-00355-w","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: An increasing body of evidence now shows that the long-term mortality of patients with sepsis are associated with various sepsis-related immune cell defects. Alternative splicing (AS), as a sepsis-related immune cell defect, is considered as a potential immunomodulatory therapy target to improve patient outcomes. However, our understanding of the role AS plays in sepsis is currently insufficient.
Aim: This study investigated possible associations between AS and the gene regulatory networks affecting immune cells. We also investigated apoptosis and AS functionality in sepsis pathophysiology.
Methods: In this study, we assessed publicly available mRNA-seq data that was obtained from the NCBI GEO dataset (GSE154918), which included a healthy group (HLTY), a mild infection group (INF1), asepsis group (Seps), and a septic shock group (Shock). A total of 79 samples (excluding significant outliers) were identified by a poly-A capture method to generate RNA-seq data. The variable splicing events and highly correlated RNA binding protein (RBP) genes in each group were then systematically analyzed.
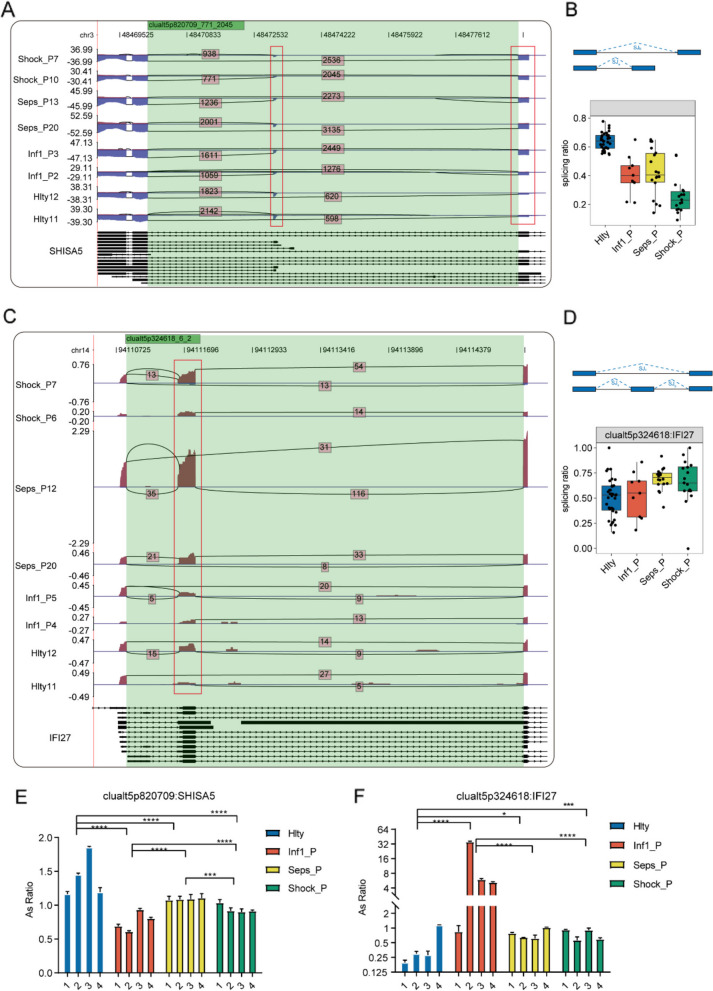
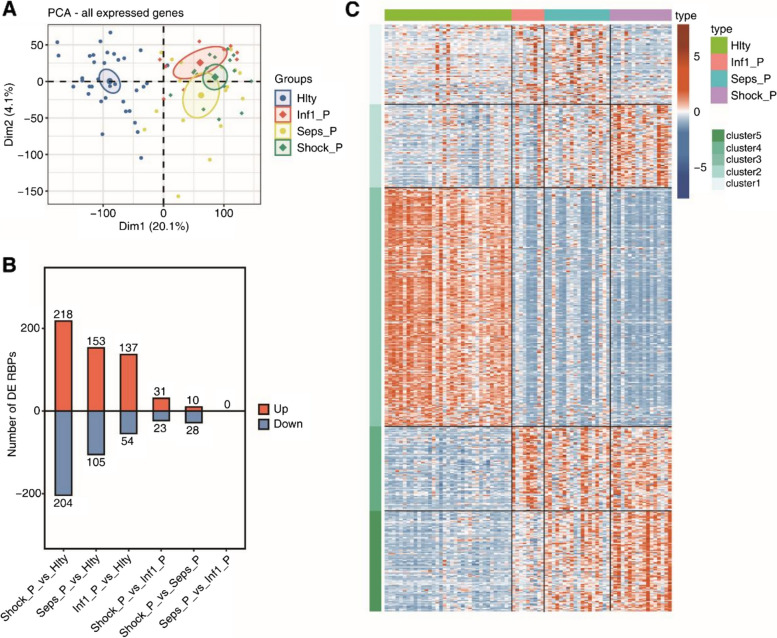
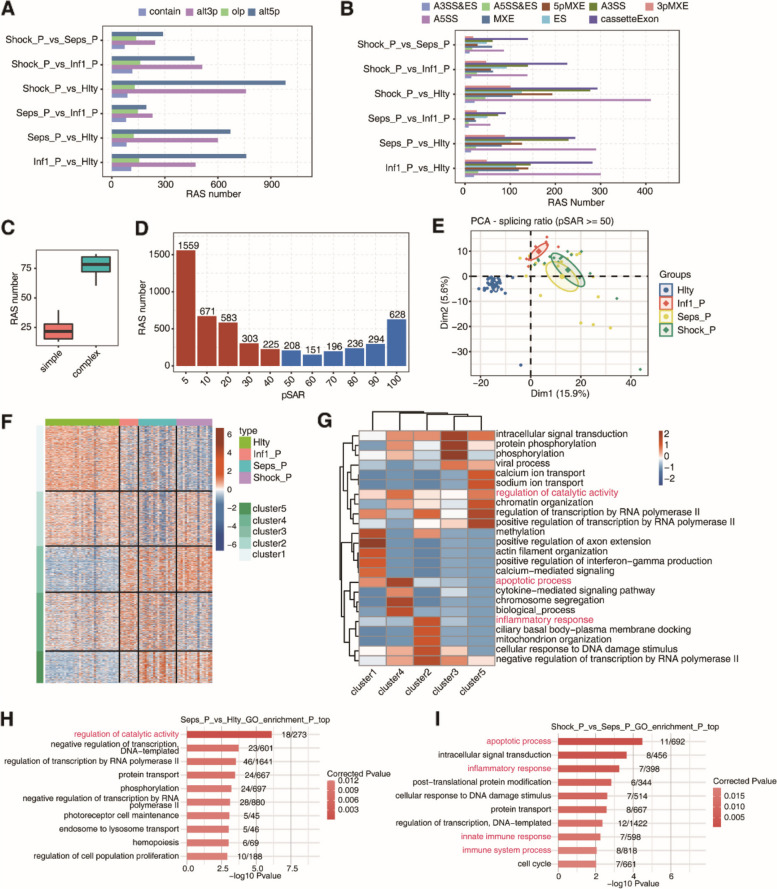
Results: For the first time, we used systematic RNA-seq analysis of sepsis-related AS and identified 1505 variable AS events that differed significantly (p <= 0.01) across the four groups. In the sepsis group, the genes related to significant AS events, such as, SHISA5 and IFI27, were mostly enriched in the cell apoptosis pathway. Furthermore, we identified differential splicing patterns within each of the four groups. Significant differences in the expression of RNA Binding Protein(RBP) genes were observed between the control group and the sepsis group. RBP gene expression was highly correlated with variant splicing events in sepsis, as determined by co-expression analysis; The expression of DDX24, CBFA2T2, NOP, ILF3, DNMT1, FTO, PPRC1, NOLC1 RBPs were significant reduced in sepsis compared to the healthy group. Finally, we constructed an RBP-AS functional network.
Conclusion: Analysis indicated that the RBP-AS functional network serves as a critical post-transcriptional mechanism that regulates the development of sepsis. AS dysregulation is associated with alterations in the regulatory gene expression network that is involved in sepsis. Therefore, the RBP-AS expression network could be useful in refining biomarker predictions in the development of new therapeutic targets for the pathogenesis of sepsis.
期刊介绍:
Journal of Inflammation welcomes research submissions on all aspects of inflammation.
The five classical symptoms of inflammation, namely redness (rubor), swelling (tumour), heat (calor), pain (dolor) and loss of function (functio laesa), are only part of the story. The term inflammation is taken to include the full range of underlying cellular and molecular mechanisms involved, not only in the production of the inflammatory responses but, more importantly in clinical terms, in the healing process as well. Thus the journal covers molecular, cellular, animal and clinical studies, and related aspects of pharmacology, such as anti-inflammatory drug development, trials and therapeutic developments. It also considers publication of negative findings.
Journal of Inflammation aims to become the leading online journal on inflammation and, as online journals replace printed ones over the next decade, the main open access inflammation journal. Open access guarantees a larger audience, and thus impact, than any restricted access equivalent, and increasingly so, as the escalating costs of printed journals puts them outside University budgets. The unrestricted access to research findings in inflammation aids in promoting dynamic and productive dialogue between industrial and academic members of the inflammation research community, which plays such an important part in the development of future generations of anti-inflammatory therapies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


